 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
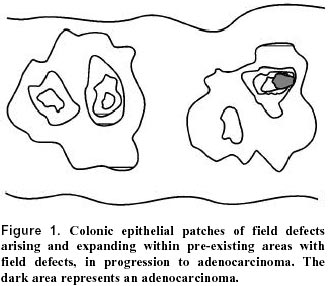
Electronic Journal of Biotechnology, Vol. 3, No. 3, December, 2000 INVITED REVIEW ARTICLE Field defects in progression to adenocarcinoma of the colon and esophagus Carol Bernstein*1, Harris Bernstein2, Claire M. Payne3 and Harinder Garewal4 1Department
of Microbiology and Immunology, College of Medicine, University of Arizona, Tucson
AZ 85724, Tel: 1-520-626-6069 Fax: 1-520-626-2100 E-mail: Bernstein3@earthlink.net
Financial support: This work was supported in part by NIEHS grant #ES06694 (Experimental Pathology Core; #ES06694 Southwest Environmental Health Sciences Center); NIH Institutional Core Grant #CA23074 to Arizona Cancer Center; NIH PPG #CA72008; Arizona Disease Control Research Commission Grant #10016: Innovite, Inc. (Tigard, Oregon) and Biomedical Diagnostics and Research, Inc. (Tucson, Arizona). Received
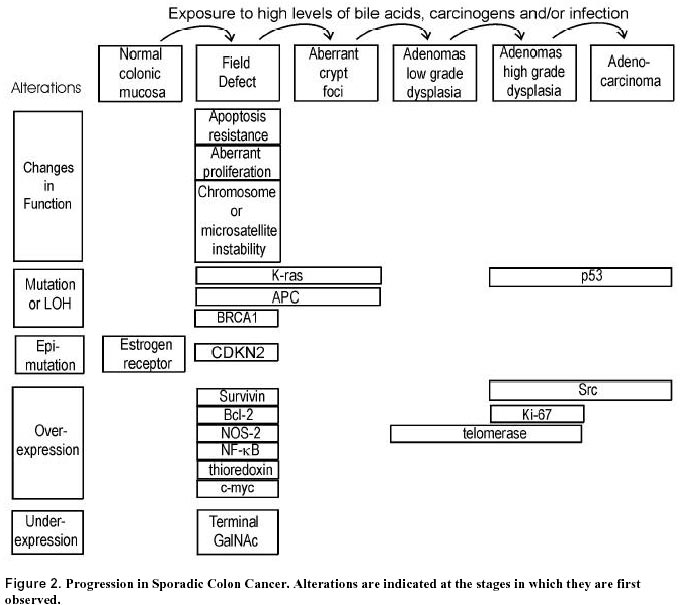
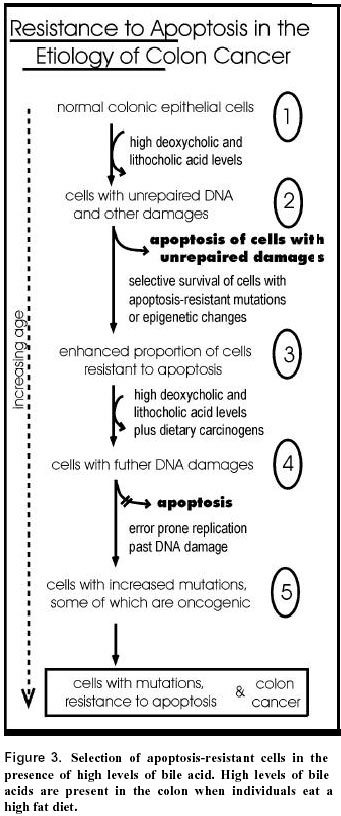
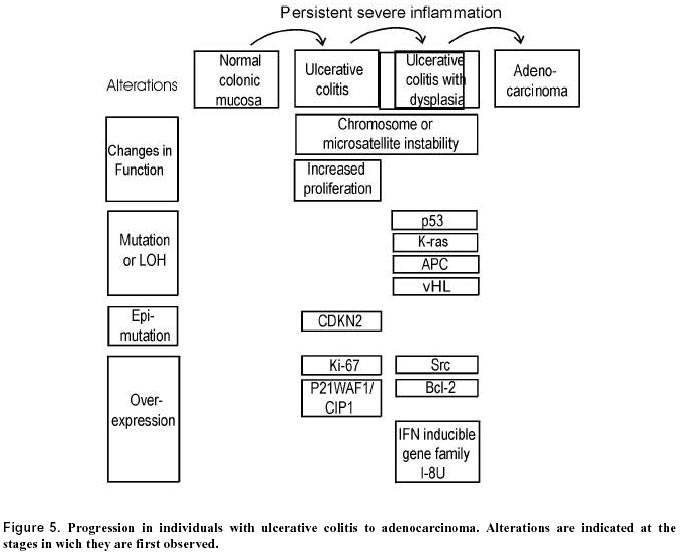
July 10, 2000 Code Number: ej00018 A field of defective tissue (field defect) appears to be an early stage in the progression to adenocarcinoma of both the colon and the esophagus. In both cases the field is comprised of cells characterized by resistance to induction of apoptosis, aberrant increased proliferation and genomic instability. Specific mutations and epimutations occur in both types of field. Also there is evidence for aberrant overexpression or underexpression of specific proteins. Most of these early molecular alterations appear to promote either enhanced apoptosis resistance, proliferation, or genomic instability. The molecular alterations that are present early are often, but not always, also present to an even greater degree in the cancer itself. Many of the important events in the development of colonic and esophageal adenocarcinoma first occur in tissues that are histologically neither dysplastic nor malignant. The identification of early molecular defects in morphologically normal appearing tissue is paramount in identifying subjects at high risk for cancer. Keywords: apoptosis resistance, barrett’s esophagus, chromosome instability, colon cancer, field effects, ulcerative colitis.A "field defect" is an area of abnormal tissue which precedes and predisposes to the development of cancer. Such abnormal fields are of interest because they give insight into the early stages of carcinogenesis and may provide biomarkers of cancer risk. The evidence for field defects varies with the type of cancer. We will focus on adenocarcinoma of the colon and esophagus, as there is substantial evidence for field defects in these two types of cancer. The term "field effects" is also commonly used in the literature. Field effect refers to changes in non-malignant tissue associated with a cancer, and is non-committal with respect to whether the field represents a beneficial response to the cancer, a neutral consequence, or a predisposing cause of the cancer. We use the term field defect to indicate our focus on tissue fields for which evidence suggests there are functional defects on the pathway to the development of cancer. In principle, the formation of a solid tumor is initiated by a series of somatically inherited changes [i.e. mutations or transmissible epigenetic events (epimutations) such as methylation of CpG islands (Baylin, 1997)]. Some or all of these changes produce a growth advantage relative to surrounding cells. Genetic instability or a mutator phenotype, arising early, may accelerate this process (Jackson and Loeb, 1998; Tomlinson and Bodmer, 1999). If, in a normal population of dividing cells within a tissue, a cell acquires a growth advantage through a mutation or an epimutation, it will tend to expand clonally at the expense of neighboring cells. Thus a patch of abnormal tissue will arise. Within this patch, a second such change may occur so that a given cell acquires a growth advantage compared to other cells within the patch, and this cell will expand clonally forming a secondary patch within the first patch. Within this new patch, the process may be repeated several more times until a malignant cell arises which clonally expands into a cancer (Figure 1). If this is the general process by which solid tumors arise, then tumors generally should be associated with, and be preceded by, a field of abnormality reflecting the succession of premalignant events. Somatic mutations or epimutations contributing to malignant progression may arise by interaction of cells with the environment (e.g. smoke components, dietary carcinogens), or by excessive production of endogenous agents causing stress (e.g. excess bile acid secretion in response to a high fat diet). Germ-line mutations in genes such as the adenomatous polyposis coli (APC) gene may also predispose individuals to cancer. In the latter case, the whole individual can be regarded as the initial predisposed field. However, whether the initial mutation or epimutation is in a somatic cell or has been transmitted through the germ line to all the cells of an individual, subsequent mutations or epimutations will likely generate a local field as in Figure 1. In general, a growth advantage may be acquired either by a decreased propensity to undergo apoptosis or by increased cell division (proliferation). Both of these types of change will not only lead to clonal expansion, but can also increase the likelihood of mutation. A reduced ability to undergo apoptosis is frequently observed, for instance in colon cancers (e.g. Bedi, et al. 1995). Normal cells which have received DNA damage, and which are unable to sufficiently repair this damage, ordinarily undergo apoptosis. This process of altruistic cell suicide is advantageous to humans since it avoids the potential of replicating DNA with a damaged template, likely to cause a mutation in the daughter DNA. Such mutations can lead to cancer. Thus a cell which is defective in the ability to undergo apoptosis in response to unrepaired DNA damage may not only have a growth advantage, but is also more likely to have excessive DNA damage and undergo mutation. In support of this concept, Kuo et al. (1999) demonstrated that overexpression of the anti-apoptotic k protein prevents benzene metabolite-induced apoptosis and attenuates the repair of oxidative DNA damage, leading to enhanced mutation among surviving cells. In the case of a clone that divides more rapidly (increased proliferation), mutations are more likely to arise over time, because, to a first approximation, mutation rate per unit time is proportional to number of cycles of DNA replication (to the extent that mutations arise as errors of replication). Given these considerations, a field of defective cells is likely to contain mutations and/or epigenetic alterations that promote either resistance to apoptosis or more frequent cell division, or both. In this review we will consider evidence for the presence of field defects, as well as specific defects that influence apoptosis, rates of cell division and mutation. Our concern here is with the early events in the carcinogenesis pathway preceding malignancy, rather than later events in the progression to metastatic cancer. Sporadic adenocarcinoma, the most common type of colon cancer, may arise by a pathway involving the following stages: normal flat mucosa, formation of a field of defective flat mucosa, aberrant crypt foci, adenoma with low grade dysplasia, adenoma with high grade dysplasia, and adenocarcinoma. These stages, along with the functional and genetic changes reported to occur in the early stages of progression to sporadic colon cancer are outlined in Figure 2. However, it is currently unclear whether all sporadic adenocarcinomas progress through each of these stages. We will now discuss some of the molecular, genetic, epigenetic and physiologic changes that characterize the field defects preceding the development of adenocarcinoma. Many of these changes probably predispose an individual to the neoplastic transformation process. Bile acids have been implicated as important etiologic factors in colon cancer (reviewed in Cheah, 1990). Bile acids induce apoptosis in colonic goblet cells at concentrations comparable to those found in fecal water after high-fat meals (Payne et al. 1995). Increased apoptosis caused by bile acids may select for cells with an apoptosis resistant phenotype as shown in Figure 3. In this figure, level (1) indicates normal colonic epithelial cells. These cells then encounter high physiologic levels of bile acids after a high fat meal. Bile acids cause DNA damage (Kandell and Bernstein, 1991; Booth et al. 1997; Venturi et al. 1997) as indicated in level (2). Most cells with DNA damage undergo apoptosis, allowing selective survival of cells with apoptosis-resistant mutations or apoptosis-resistant epigenetic changes as shown on level (3). Upon further DNA damages, as shown in level (4), this population of apoptosis-resistant cells will be blocked in apoptosis. The apoptosis-resistant cells will allow error-prone replication past the DNA damages, resulting in cells with further mutations as shown in level (5). We have found that goblet cells within the normal-appearing (non-tumorous) portion of the colonic epithelium of colon cancer patients are, on average, more resistant to bile acid-induced apoptosis than are colonic goblet cells from individuals with neoplasia-free colons (Payne et al. 1995; Garewal et al. 1996; Bernstein et al. 1999). Furthermore, when pairs of biopsies were taken from the cecum, 40cm and 20cm from the anal verge, we observed that in patients with a history of colon cancer apoptosis resistance tended to have a patchy distribution occurring both close to and at considerable distances away from the tumor (Bernstein et al. 1999). Kozoni et al. (2000) studied a mouse model in which colon cancer is induced by dimethylhydrazine (DMH) and the secondary bile acid, lithocholic acid (LCA), is used as a promoter. They found that when DMH was injected once a week for 28 days, upon sacrifice, there was a nearly 50-fold increase in apoptosis in the basal third of colonic crypts. However, when the mice received the same DMH treatment but also received twice daily intrarectal administration of LCA, there was complete suppression of apoptosis at sacrifice (after 28 days of treatment). The enhanced apoptosis observed with DMH treatment alone was interpreted as an appropriate response by the colon to a damaging agent to eliminate cells that ought not to survive and proliferate. In contrast, the complete suppression of apoptosis by LCA in the presence of the carcinogen was interpreted to facilitate the development of cancer. Apoptosis resistant fields may arise if gene products which inhibit apoptosis are overexpressed and/or gene products which promote apoptosis have reduced expression. As discussed below, gene products which inhibit apoptosis include Bcl-2, Survivin, NF-kB, NOS2 and thioredoxin, whereas gene products which promote apoptosis include APC and P53. Bcl-2 is one of the most biologically relevant inhibitors of apoptosis (e.g. Kuo et al. 1999). Aberrant overexpression of Bcl-2 is frequently observed in colorectal adenomas and in carcinomas (Hague et al. 1994; Bosari et al. 1995; Bronner et al. 1995). Bronner et al. (1995) reported that there was frequent abnormal expression of Bcl-2 in nondysplastic epithelium surrounding dysplastic lesions, suggesting that altered expression occurs before the development of morphological dysplasia. Morphologically nondysplastic epithelium directly contiguous to a malignant or dysplastic lesion often showed abnormal Bcl-2 expression throughout the full length of the crypt-villus axis. Proceeding away from the lesion, this expression pattern gradually diminished to involve only the crypt base (the normal pattern of expression). Ohmori et al. (1999) also found increased expression of Bcl-2 in mucosa neighboring the tumor, as well as increased correlated expression of several neurohormonal polypeptides. Expression of the apoptosis inhibitor gene, Survivin, in colon tumors, predicts poor survival due to subsequent recurrent colorectal carcinoma (Sarela et al. 2000). Approximately half the patients with Survivin positive tumors, but none of the patients with Survivin negative tumors, had normal appearing mucosa which also expressed this gene. This suggests that a Survivin positive tumor arose in a Survivin positive field of normal appearing mucosa, and that other areas of normal appearing mucosa in the same patient also have this field defect. NF-kB is a redox sensitive transcription factor. We found that inhibition of NF-kB sensitizes cells to bile acid induced apoptosis, indicating that this protein protects against apoptosis (Payne et al. 1998). One downstream effector of NF-kB is nitric oxide synthase (NOS2). We have also observed that inhibition of NOS sensitizes cells to bile acid induced apoptosis and thus the product of NOS, nitric oxide, may be part of a signaling pathway that is responsible for apoptosis resistance (Payne et al. 1999; Washo-Stultz et al. 1999). Thioredoxin, a protein overexpressed in human cancer, also inhibits apoptosis (Baker et al. 1997). Recently, using immunohistochemical staining, we found that NOS2, NF-kB and thioredoxin are aberrantly expressed in the flat mucosa of individuals with colon neoplasia, and that this aberrant expression is correlated with loss of apoptosis competence (Payne et al. 2000). The aberrant expression of these proteins was represented by decreased expression at the surface epithelium, up-regulation in the crypt zones of differentiation and proliferation, and increased expression in interstitial immune cells. The APC gene normally functions to promote apoptosis. Inactivation of APC in colonic epithelium has been implicated in neoplastic transformation (Morin et al. 1996). An inherited defect in the APC gene is the first step for colorectal polyp formation and the development of colon cancer in familial adenomatous polyposis (FAP) patients (Ichii et al. 1992). A similar defect in APC is also detected in 60% of spontaneous colorectal cancers and 63% of adenomas (Powell et al. 1992) and in 4.6% of aberrant crypt foci (Smith et al. 1994), indicating that APC mutation can be an early step in sporadic colon cancer. These results suggest that loss of the pro-apoptotic APC protein contributes to the generation of an apoptosis-resistant field that predisposes to development of neoplasia. Bedi et al. (1995) found that individuals with familial adenomatous polyposis (FAP), due to an inherited defect in the APC gene, have normal-appearing flat colonic mucosa in which cells have a reduced ability to undergo stress-induced apoptosis compared to individuals with no history of colon neoplasia. They also found that transformation of colorectal epithelium to carcinomas was associated with a progressive inhibition of apoptosis. Point mutations in gene p53 in combination with loss of heterozygosity (LOH) for chromosome 17p (which includes the p53 gene) seem to occur only as late events in colorectal tumorigenesis after formation of adenomatous polyps (Figure 2) (Baker et al. 1990). This contrasts with the anti-apoptotic proteins Bcl-2, NF-kB, Survivin, NOS2 and thioredoxin which appear to be overexpressed early in the progression to colon cancer based on their elevated levels in the flat mucosa of patients with a history of colon cancer, and mutations in the pro-apoptotic gene APC which also occur early (Figure 2).
Scalmati and Lipkin (1993) reviewed numerous studies on cell proliferation pattern in individuals with colon adenomas or cancer where incorporation of tritiated thymidine or bromodeoxyuridine into DNA (labeling index) was mainly used as the measure of cell proliferation. They noted that expansion of the proliferative compartment is present along the entire colon in individuals at increased risk, no matter where the lesion is. The proliferative compartment of normal colorectal mucosa of individuals at low risk for colorectal cancer is located in the lower two-thirds of the colonic crypts. In patients at increased risk the proliferative compartment is expanded towards the top of the crypts. However, since proliferative rates in colonic crypts are highly variable in the same individual and can be influenced by diet, its value as a biomarker for colon cancer risk is doubtful. Genetic and epigenetic changes in gene expression that enhance proliferation in the progression to adenocarcinoma In addition to the genetic alterations that reduce apoptosis, discussed above, other alterations occurring early in progression to colon adenocarcinoma appear to enhance cell proliferation. These include increased expression of C-myc and K-ras and decreased expression of estrogen receptor. C-myc, the normal product of the myc proto-oncogene, is a nuclear transcription factor. C-myc is overexpressed in colon cancer cells and was also found to be overexpressed in non-neoplastic mucosa adjacent to tumors (Visca et al. 1999). Since C-myc is normally repressed by wild-type APC (He et al. 1998), the presence of APC mutations may account for C-myc overexpression. K-ras oncogene is a guanine nucleotide-binding protein with GTPase activity that is involved in signal transduction. Non-neoplastic mucosa and tumor tissues from 70 patients with colorectal cancer were analyzed by the enriched polymerase chain reaction for mutations in codon 12 of the K-ras gene (Minamoto et al. 1995). In 18-25% of colorectal cancer patients, mutant K-ras was identified in the apparently normal mucosa. Zhu et al. (1997) reported that K-ras codon 12 mutations occur in a high frequency (53.8%) in histologically normal mucosa adjacent to tumors of patients with K-ras mutation-positive colorectal cancer. K-ras mutations have also been identified in aberrant crypt foci (Smith et al. 1994; Yamashita et al. 1995). These results suggest that cells with K-ras mutations might reflect a field defect predisposed to further progression to cancer. CpG islands are located in the promoter region of about 60% of genes and are ordinarily free of methylation, regardless of the expression state of these genes (except when these genes are silenced within the inactive X chromosome of women or in the transcriptionally silent copy of parentally imprinted genes). In neoplastic cells, however, some genes appear to acquire de novo methylation in their CpG islands. CpG island methylation is an epigenetic modification of DNA (epimutation) known to correlate closely with silencing of gene transcription. Issa et al. (1994) have reported CpG island methylation occurs in the estrogen receptor (ER) gene in a subpopulation of cells of the colonic mucosa, and this CpG island methylation increases as a direct function of age. This change was also found in virtually all cells in all colorectal tumors examined, and in both large and small adenomatous polyps. Additionally Issa et al. (1994) presented evidence that expression of the unmethylated ER gene results in growth inhibition of colon carcinoma cells. Thus the estrogen receptor appears to play a role in growth regulation of colonic cells and its transcriptional repression by CpG island methylation could contribute to deregulated growth of these cells. The methylation-associated inactivation of the ER gene in aging colorectal mucosa could be one of the earliest events that predisposes colorectal mucosa to sporadic tumorigenesis with age. Loss of cell cycle control is among the defining features of tumor cells, and inactivation of cell cycle inhibitors is a common mechanism by which this loss occurs. The CDKN2 gene product, p16INK4a, is a critical cell cycle inhibitor that binds to the complex of cyclin D and cyclin-dependent kinase 4 or 6 (CDK4 or CDK6) to repress its ability to phosphorylate the retinoblastoma protein. Binding of p16INK4a normally inhibits the kinase activity, thereby leaving the RB protein underphosphorylated and consequently promoting cell cycle arrest. In contrast, loss of p16INK4a promotes cell proliferation, and mutation or inactivation of the CDKN2 gene is found in many different types of human cancers. Gonzalez-Zulueta et al. (1995) reported extensive methylation of CpG islands of the CDKN2 promoter in the normal-appearing flat mucosa of the colon in 60% of patients with colon cancer.
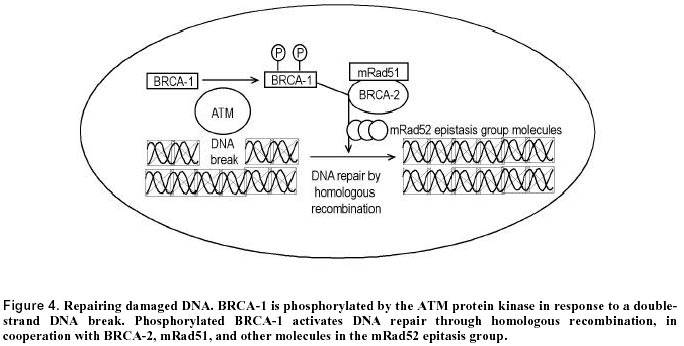
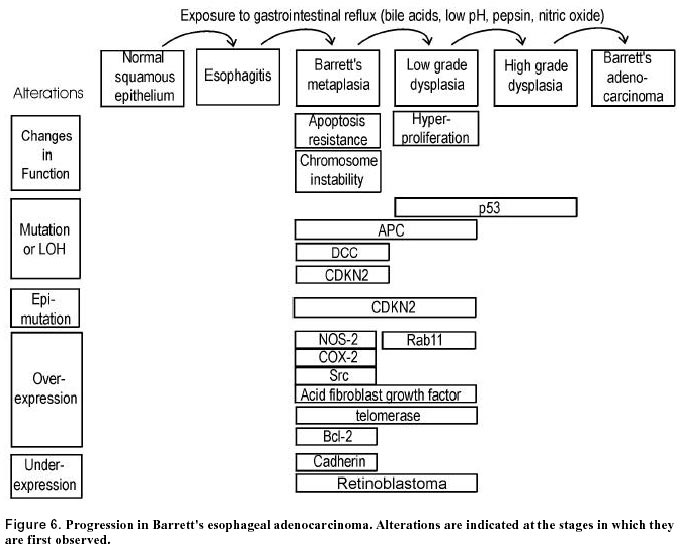
Recently we found that the flat colonic mucosa of colon cancer patients have field defects characterized by dedifferentiation (Holubec et al. 2000). Tissues from individuals without neoplasia showed normal differentiation in 85% of samples as evidenced by histochemical staining with the lectin Dolichos biflorus agglutinin, which reacts with the N-acetyl galactosamine residues of the mucin of mature goblet cells. Tissue from individuals with tubulovillous adenoma showed 63% normal differentiation, and tissue from colon cancer patients showed only 41% normal differentiation, implying frequent areas of field defect. Premalignant changes in gene expression occurring in adenomatous polyps Adenomas are benign polyps that may progress to malignancy, and can thus be regarded as fields of defective tissue. However, most adenomas do not progress to cancer. Therefore, additional alterations in adenomas promote proliferation and predispose them to cancer. Telomerase is a ribonucleoprotein that synthesizes telomeric DNA onto chromosomal ends. Expression of telomerase is often associated with cellular immortality and oncogenesis. Telomerase has been found to be reactivated or up-regulated in most cancers. Fang et al. (1999) detected telomerase activity in 89% of colorectal carcinomas, in 50% of colonic adenomas, but not in normal colorectal mucosa. Yan et al. (1999) found telomerase activity in all colonic adenomas showing high grade dysplasia and in adenocarcinomas, whereas activity was detected in only 20 percent of low grade adenomas and in none of the normal colonic mucosal tissue samples from individuals without neoplasia. Thus, telomerase activation is often associated with progression from low-grade to high-grade dysplasia in adenomas. Yoshida et al. (1999) also reported results indicating that telomerase activation gradually increases during the course of colorectal carcinogenesis. Ki-67, a proliferation factor, was measured immunohistochemically, and found to be associated with adenomas having high grade dysplasia (Visca et al. 1999). Src proto-oncogene encodes a cytoplasmic nonreceptor protein-tyrosine kinase. Src tyrosine kinase is elevated in colon cancer (Bolen et al. 1987; Cartwright et al. 1989) and in colonic polyps of high malignant potential (Talamonti et al. 1993). Chromosome instability (CIN) and microsatellite instability (MIN) Substantial evidence, reviewed by Lengauer et al. (1997), indicates that nearly all cancers are characterized by genetic instability, but that the instability exists at two distinct levels. In a small subset of tumors the instability is observed at the nucleotide level, resulting in base substitutions or deletions or insertions of a few nucleotides. In most other cancers the instability is observed at the chromosome level, resulting in losses and gains of whole chromosomes (aneuploidy) or large portions thereof, as well as chromosomal translocations, insertions and deletions larger than a few nucleotides. Colorectal cancers fall into two groups with regard to instability. Roughly 13% of colorectal cancers contain defects in mismatch repair, leading to instability at the nucleotide level. This form of instability is detected as microsatellite instability (MIN). Microsatellites are simple repetitive sequences of DNA that are scattered throughout the genome. These sequences are stably inherited, vary from individual to individual, and have a relatively low mutation rate. Instability within these sequences is a marker of genome-wide mutations and DNA repair deficiencies. MIN has been detected in cancers associated with the hereditary nonpolyposis colon cancer (HNPCC) syndrome, but also in tumors that acquired a mismatch repair defect in the somatic line. The majority of colorectal cancers show chromosomal instability (CIN). Both types of instability appear to arise early during colorectal tumorigenesis (Figure 2). HNPCC syndromes are defined by the occurrence of colon cancer in at least three first-degree relatives representing at least two generations, and the development of cancer in one of these individuals before age 50. Germline mutations in any one of four homologues of bacterial DNA mismatch repair genes (hMLH1, hMSH2, hPMS1 or hPMS2) result in HNPCC. Disruption of these genes causes an increase in MIN. In this form of instability, stretches of tandemly repeated dinucleotide and trinucleotide sequences are found to be expanded or contracted in tumors from HNPCC patients. More than 50% of colon adenocarcinomas having the microsatellite mutator phenotype were found to have a frameshift mutation in a tract of eight deoxyguanosines within BAX, a gene that promotes apoptosis (Rampino et al. 1997). Such adenocarcinomas were also found to have mutations in the pro-apoptotic APC gene which were disproportionately of the frameshift type (Huang et al. 1996). Mutations in APC, BAX and other pro-apoptotic genes may account, in part, for the observed apoptosis resistance previously described. The molecular basis for CIN, as distinct from MIN, is just beginning to be explored. Stoler et al. (1999) have found chromosomal alterations in sporadic colorectal cancer to be considerably more abundant than expected, with the mean number of genomic events per carcinoma totaling approximately 11,000. Colonic polyps early in the tumor progression pathway showed similar numbers of events. These results indicate that genomic destabilization is an early step in sporadic tumor development. p53 mutations were shown to be unlikely to initiate or promote genomic instability in sporadic colorectal tumors (Kahlenberg et al. 1996). Lengauer et al. (1997) showed that an abnormal number of chromosomes in a cell, in and of itself, cannot account for the high level of CIN found in aneuploid colorectal cancers. An attractive explanation for CIN is that it arises in cells that enter mitosis before DNA damages are repaired because of defects in DNA repair or checkpoint genes. In particular, defective ability to repair double-strand breaks through the process of recombinational repair may be implicated in CIN. Thus, defects in genes involved in recombinational repair, such as BRCA1 and BRCA2, are good candidates for mediating CIN. Garcia-Patino et al. (1998) reported that 49% of colon cancers have LOH for chromosomal region 17q21, the region which contains the BRCA1 gene. A preliminary study of the incidence of BRCA1 and BRCA2 mutations in Ashkenazi colorectal cancer patients indicated elevated rates of these mutations (Drucker et al. 2000). These findings suggest that defective BRCA1 and BRCA2 genes may play an important role in the development of colon cancer. Venkitaraman (1999) has indicated the likely roles of BRCA-1 and BRCA-2 proteins in DNA repair by homologous recombination. Figure 4 illustrates a pathway suggested by Venkitaraman in murine cells. It is clear, from this figure, that a defect in BRCA1 or BRCA2 would result in increased numbers of double strand breaks (DSBs). It was found (Xu et al. 1999) that a targeted deletion within exon 11 of BRCA1 in mouse embryonic fibroblast cells indeed generates extensive chromosomal abnormalities, a characteristic of CIN. Increased numbers of DSBs would ordinarily trigger apoptosis, unless the cells were also apoptosis resistant. Thus, cells subject to DSBs, and CIN, would be under strong selective pressure to become defective in apoptosis. The possibility of a predisposing field arising from infection Swidsinski et al. (1998) investigated the association between intraepithelial infection by bacteria, mainly Escherichia coli, and colorectal cancer. They reported intracellular E. coli in colonic tissue of 87% of patients with adenoma and carcinoma and in none of the mucosa of neoplasia-free individuals. To estimate intracellular E. coli concentrations, a gentamicin protection assay was used involving plating and colony formation. High bacterial counts were seen not only in tumors, but in both adjacent and distant normal tissue and in biopsy specimens from relapse-free patients within 3 years after partial colectomy. Therefore, a local effect of tumor-damaged tissue is unlikely. Although it is not clear whether intracellular E. coli colonization of the colonic mucosa is primary or secondary to the malignant pathology, these findings raise the possibility that colon cancer arises in a predisposed field of infected epithelium. JC virus is a polyoma virus that commonly infects humans. JC DNA fragments were detected both in malignant and in normal, adjacent colorectal epithelium from surgical resection specimens (Laghi et al. 1999). A larger number of viral copies was present in cancer cells than in the non-neoplastic colon cells. Further, Ito et al. (1993) isolated Inoue-Melnick virus from colorectal carcinomas as well as from the normal appearing mucosa of cancer patients, whereas individuals without colorectal cancer had no virus in their colonic mucosa. Aberrant expression of specific proteins in ulcerative colitis Chronic ulcerative colitis (UC) is an inflammatory disease associated with an increased incidence of colon cancer. UC appears to be caused by excessive immune reactivity of the gut wall, possibly due to antibody mediated hypersensitivity (MacDonald et al. 2000). Persistent severe inflammation in colonic mucosa is thought to lead to the development of colon cancer in patients with UC. This might be mediated by increased mucosal concentrations of reactive oxygen species that cause tissue damage (Sedghi et al. 1993). Adenocarcinoma in UC presumably develops from dysplastic precursor lesions, which include flat dysplasia and polypoid (polyp-like) dysplasias. The changes occurring in UC leading to colon cancer are outlined in Figure 5. The presence of mutations or LOH of the pro-apoptotic genes p53 and APC, and overexpression of anti-apoptotic gene bcl-2 in the flat mucosa of patients with UC suggest that there may be a defect in apoptosis. This may be brought about by a higher rate of apoptosis (Arai et al. 1999) which may select for an apoptosis resistant phenotype. p53 mutations are common in areas of dysplasia and in carcinomas in humans with UC (Yin et al. 1993; Fogt et al. 1998a). In contrast to sporadic colorectal carcinomas, ulcerative colitis-associated neoplastic progression appears to involve p53 inactivation at a relatively early stage. LOH of the APC gene in UC was noted in high grade dysplasia with associated cancer, but not in cases of nondysplastic epithelium or in high grade dysplasia alone (Fogt et al. 1998b). Bcl-2 expression was found to be increased in areas of flat dysplasia in ulcerative colitis but not in non-dysplastic ulcerative colitis tissue (Bronner et al. 1995). An increase in proliferation in active UC is indicated by an increase in mitotic index, p21WAF1/CIP1 labeling index and Ki-67 labeling index (Arai et al. 1999). A shift of the proliferative compartment towards the top of the crypt has been found in the non-tumerous mucosa of individuals with chronic ulcerative colitis (reviewed in Scalmati and Lipkin, 1993). K-ras mutations are also present in dysplastic regions (Anderson et al. 1999). Furthermore Src activity is elevated in dysplastic epithelia of UC (Cartwright et al. 1994). Src activity is highest in malignant and severely dysplastic epithelia, and 6-10-fold higher in mildly dysplastic compared to nondysplastic epithelia. Thus, Src activity is raised in dysplastic UC epithelia, which is at increased risk for developing cancer. p16INK4a is transcriptionally silenced as a consequence of hypermethylation of CpG islands in patients with long standing UC (Hsieh et al. 1998). As discussed above, decrease in p16INK4a promotes cell proliferation. To clarify the mechanism of carcinogenesis from chronic inflammation in colonic mucosa, Hisamatsu et al. (1999) isolated cDNA fragments corresponding to mRNAs that are differentially expressed between UC-associated cancer tissue and mildly inflamed mucosa. On this basis, they identified interferon-inducible gene family 1-8U as being strongly expressed in UC-associated cancers, but not in flat mucosa. Furthermore, this gene was also strongly expressed in severely inflamed colonic mucosa of UC without colitis-associated cancer. The function of the 1-8U gene is currently unknown. Von Hippel-Lindau (vHL) disease is a hereditary tumor syndrome characterized by predisposition for bilateral and multi-centric hemangioblastoma in the retina and central nervous system, pheochromocytoma, renal cell carcinoma, and cysts in the kidney, pancreas, and epididymis. LOH of the vHL gene occurs in, and also nearby areas of dysplasia in UC (Fogt et al. 1998a). Ulcerative colitis tissue shows weak, if any, telomerase activity (Yoshida et al. 1997). Brentnall et al. (1996) reported MIN in 50% of UC patients whose colonic mucosa was negative for dysplasia, 46% of those with high-grade dysplasia, and 40% of those with cancer, whereas normal colonic tissue showed no evidence of MIN (Figure 5). In some of the patients with cancer or high grade dysplasia, the MIN occurred in histologically negative tissue remote from the site of the cancer or dysplasia. In UC patients with high-grade dysplasia or cancer, CIN is found not only in areas of dysplasia, but is also present throughout the colon (Rabinovitch et al. 1999). These findings suggest that widespread MIN and/or CIN precedes dysplasia and cancer in UC and may reflect the inability of DNA repair mechanisms to compensate for the stress of chronic inflammation. Adenocarcinoma of the esophagus Barrett’s esophagus (BE) is a lesion resulting from chronic gastroesophageal reflux disease (GERD) in which the stratified squamous epithelium of the esophagus is replaced by metaplastic columnar epithelium that predisposes to the development of esophageal adenocarcinoma. At the gross level, Barrett’s mucosa usually appears as a well-defined area, with irregular margins, consisting of salmon-pink, velvety mucosa similar to the adjacent gastric mucosa (Coppola and Karl, 1999). However, biopsy is required for diagnosis. Most investigators believe that the increased cancer risk is associated with specialized columnar epithelium (SCE) or intestinal metaplasia. At the histological level, its defining feature is the presence of goblet cells, similar to those of the epithelium of the small intestine and colon. These goblet cells are found in the surface epithelium and in crypts. The sequence of histological events in the progression from BE to adenocarcinoma, is considered to be (1) Barrett’s metaplasia; (2) low grade dysplasia; (3) high grade dysplasia and (4) invasive adenocarcinoma. About 6% to 15% of individuals with chronic GERD develop BE. GERD exposes the lower esophagus to bile acids from the duodenum and acid/pepsin from the stomach which, in combination, may be the cause of the development of BE. Oxidative damage appears to play a role in this process in humans (Olyaee et al. 1995; Wetscher et al. 1995) as well as in a rodent model of esophageal adenocarcinoma (Chen et al. 2000). In a rat model of the regulatory mechanism of acid secretion in the stomach after damage with the bile acid taurocholate, endogenous nitric oxide was found to down-regulate acid production (Takeuchi et al. 2000). Approximately 0.5% to 1.0% of patients with Barrett’s metaplasia develop adenocarcinoma, one of the most lethal of all cancers (Reynolds et al. 1999). The adenocarcinoma ordinarily arises in a field of Barrett’s metaplasia (Coppola and Karl, 1999). BE is perhaps one of the best examples of a field defect as it is clearly distinguishable from the usual, normal squamous epithelium. At present, assessment of grade of dysplasia, using endoscopic biopsies, is the best marker available to identify high risk of progression to adenocarcinoma. However, subjectivity limits the classification into discrete grades of dysplasia (Reynolds et al. 1999). Therefore, numerous workers have sought molecular markers as more reliable indicators of cancer risk. Although no particular molecular marker has yet gained widespread acceptance, this work has given insight into the functional defects and genetic events occurring within the abnormal field leading to esophageal adenocarcinoma (see Figure 6 for summary). Katada et al. (1997) observed that inhibition of apoptosis occurs early in the metaplasia-dysplasia-carcinoma sequence of BE. Wetscher et al. (1998) also reported that apoptosis was low in Barrett’s epithelium compared to adjacent squamous epithelium. Whittles et al. (1999) reported a decrease in apoptosis in the upper crypt and luminal surface in dysplasia and adenocarcinoma compared with Barrett’s metaplasia. Katada et al. (1997) reported overexpression of the bcl-2 anti-apoptotic proto-oncogene in 72% of Barrett’s metaplasia samples and in 100% of Barrett’s low grade dysplasia, although Goldblum and Rice (1995) did not find Bcl-2 overexpression in these tissues. We have found that, within the region of Barrett’s metaplasia, the goblet cells are highly resistant to bile acid induced apoptosis compared to the goblet cells of the normal colon epithelium (unpublished results), suggesting that chronic reflux and consequent exposure to bile acids may select for resistance to bile acid induced apoptosis. Thus, it appears that relatively reduced apoptosis capability is a characteristic of the field of Barrett’s metaplasia and that this capability is further reduced in the progression to dysplasia and adenocarcinoma. Aberrant expression of pro-apoptotic P53 and APC, and of anti-apoptotic NOS-2 and COX-2 There is evidence for aberrant expression of proteins P53, APC, NOS-2 and COX-2 in the metaplastic and dysplastic epithelial field preceding development of esophageal adenocarcinoma that would tend to reduce apoptosis capability. LOH for the p53 gene occurs in Barrett’s metaplasia and dysplasia adjacent to adenocarcinoma of the esophagus (Dolan et al. 1999; Galipeau et al. 1999). This finding suggests that LOH at the site of the p53 gene occurs before the esophageal adenocarcinoma develops. Mutations of the p53 gene are associated with an increased half-life of the P53 protein, leading to its accumulation. This accumulation can be detected by specific antibodies and immunohistochemical staining, and thus can be used to identify cells with a presumed p53 mutation. Overexpression of P53 has been found in over 50% of adenocarcinomas arising in BE (e.g. Blount et al. 1991; Flejou et al. 1993; Younes et al. 1993; Casson et al. 1995). Rioux-Leclercq et al. (1999) found P53 overexpression in only 7% of tissues with low grade dysplasia, but in 60% of tissues with high grade dysplasia, and in 85% of adenocarcinomas. Similar results were obtained by Younes et al. (1993) and Ramel et al. (1992). However, Younes (1997) reported overexpression of P53 in 36% of areas with low grade dysplasia and this was associated with increased probability of progression to high grade dysplasia. Also Cawley et al. (1998) presented evidence that BE and esophageal cancer can develop P53 antibodies that may predate the clinical diagnosis of malignancy. It thus appears that overexpression of P53 is an early event in the progression to esophageal adenocarcinoma. LOH for the pro-apoptotic APC gene occurs in Barrett’s metaplasia and dysplasia adjacent to an adenocarcinoma, and also in the adenocarcinoma itself (Zhuang et al.1996; Dolan et al. 1999) The two anti-apoptotic proteins, inducible nitric oxide synthase (NOS2) and cyclooxygenase-2 (COX-2), exhibited elevated expression in both BE and associated adenocarcinoma (Wilson et al. 1998; Shirvani et al. 2000), and thus may be significant at early times in the metaplasia-dysplasia-adenocarcinoma sequence (Figure 6). Rioux-Leclercq et al. (1999) measured cell proliferation by histochemistry using antibody against the Ki-67 nuclear antigen. They found an increase in proliferation in the progression from low-grade dysplasia to high-grade dysplasia to carcinoma. Similarly, Whittles et al. (1999) found an increase in proliferation in dysplastic and carcinomatous tissue compared with metaplastic tissue. Thus it appears that the field of aberrant cells in which esophageal cancer arises experiences a progressive increase in proliferation, as well as a progressive decrease in apoptosis. Aberrant expression of proteins involved in proliferation LOH of chromosome 9p21 (which includes the CDKN2 gene) and CDKN2 mutations occur as early lesions in diploid cells before the development of aneuploidy and cancer during neoplastic progression in BE (Barrett et al. 1996; Galipeau et al. 1999). However only a minority of Barrett’s adenocarcinomas with 9p21 LOH have a somatic mutation in the other CDKN2 gene on the homologous chromosome. A common alternative mechanism of CDKN2 inactivation appears to be hypermethylation of the CDNK2 promoter (Wong et al. 1997) Src is activated in BE and esophageal adenocarcinoma (Kumble et al. 1997). Src activation occurs before development of dysplasia or carcinoma (Figure 6). Different regions of BE esophagus from the same patient showed heterogeneity in Src activity compared with the uniform Src activity observed in different regions of the normal esophagus. This implies the field of Src defect is patchy. Acid fibroblast growth factor belongs to a family of polypeptides that are involved in differentiation and cellular proliferation. This factor is accumulated in the progression from metaplasia to adenocarcinoma (Soslow et al.1997). Telomerase activation is frequently associated with cellular immortality and carcinogenesis (Perrem and Reddel, 1999). Morales et al. (1998) found that 1) Barrett’s epithelium may contain a population of immortalized cells, 2) a marked increase in the level of telomerase RNA accompanies the transition form low grade to high grade dysplasia, and 3) high levels of telomerase RNA accompany the development of esophageal adenocarcinoma in the vast majority of cases. Lord et al. (2000) also found that telomerase expression is increased early in the Barrett’s metaplasia-dysplasia-adenocarcinoma sequence. Telomerase expression was also significantly higher in histologically normal squamous epithelium of the esophagus from cancer patients than in normal squamous epithelium of the esophagus from patients with no cancer, suggesting that a widespread defective field is present in the esophagus of patients who develop Barrett’s cancer. Cadherins are a family of transmembrane glycoproteins involved in calcium dependent intercellular adhesion. One of the subclasses of cadherins, E-cadherin appears to be the prime mediator of cell-cell interactions in epithelial cells. Down-regulation of E-cadherin appears to favor the processes of invasion and metastasis. E-cadherin was found to be significantly lower in Barrett’s epithelium compared to normal esophageal epithelium and even lower in esophageal adenocarcinoma (Swami et al. 1995) Rab11 is a small GTP-binding protein which is involved in intracellular vesicle membrane processing. Ray et al. (1997) reported that perinuclear staining for Rab11 is associated specifically with low grade dysplasia of Barrett’s epithelium. LOH for the Deleted in Colon Cancer (DCC) gene occurs in Barrett’s metaplasia and also in the adenocarcinoma itself (Zhuang et al. 1996; Dolan et al. 1999). Although it was once thought that this gene is involved in goblet cell differentiation, recent results show that DCC is not selectively involved in the mucosecretory differentiation pathway and that it is neither sufficient nor essential for normal intestinal differentiation (Fabre et al. 1999). Retinoblastoma protein (Rb), in its activated unphosphorylated form, sequesters transcription factors needed for G1-S progression (Weinberg, 1995). Excess proliferation is frequently associated with deregulation of the Rb pathway through loss of Rb. Loss of Rb staining was observed as metaplasia progressed to dysplasia and carcinoma in the esophagus (Coppola et al. 1999). Garewal et al. (1989) showed that BE is frequently characterized by clonal proliferation of karyotypically abnormal cells. Walch et al. (2000) characterized cytogenetic alterations in Barrett’s adenocarcinoma and its premalignant stages. The average number of detected chromosomal imbalances increased from 7.0 in Barrett’s metaplasia to 10.8 in low grade dysplasia to 13.4 in high grade dysplasia and 13.3 in adenocarcinoma. The occurrence of common chromosomal alterations in premalignant lesions and adjacent carcinomas suggested a process of clonal expansion, whereas differences between these adjacent tissues suggested that the clonal expansion was accompanied by genetic divergence. The evidence summarized in this review indicates that adenocarcinomas of both the colon and esophagus arise in a predisposed defective field. The most common setting in which a colonic adenocarcinoma likely arises is a field of flat mucosa characterized by a pattern of apoptosis resistance, aberrant proliferation, and CIN in which sporadic aberrant crypt foci give rise to adenomas, which in turn give rise to an adenocarcinoma. Certain features appear to be common to this type of field progression and the metaplasic-dysplastic field progression in which esophageal adenocarcinoma arises. These features include: (1) reduced apoptosis capability, possibly resulting, in part, from chronic exposure to elevated levels of bile acids; (2) aberrant proliferation; (3) CIN; (4) APC mutation as an early event; (5) aberrant over-expression of NOS-2, Src, Bcl-2 and telomerase; (6) hypermethylation and silencing of the CDKN2 promoter. There are also some significant differences between the two types of field. These include: (1) K-ras mutations are common in colonic adenomas and adenocarcinomas, but were not found in BE (Meltzer et al. 1990). (2) the absence of adenomatous polyps as precursor lesions in the Barrett’s progression contrasts with the role of polyps in sporadic colonic adenocarcinoma. (3) p53 mutation occurs earlier in progression to esophageal adenocarcinoma than in progression to sporadic colonic adenocarcinoma. Another setting in which colonic adenocarcinoma arises is in ulcerative colitis. Ulcerative colitis and esophagitis have in common that both (1) are fields of inflammation predisposing to adenocarcinoma; (2) tend to have mutations in p53, APC and K-ras. (3) have overexpression of Src tyrosine kinase and Bcl-2; (4) have hypermethylation of the CDKN2 promoter. In the Introduction, we argued that field defects are a logical consequence of the assumption that cancers arise by a sequence of mutations and/or epimutations each of which confers a growth advantage. The defective field, by this logic, should reflect the predisposing mutations and epimutations that precede the development of the malignancy. We have reviewed here the relevant evidence for the existence of field defects predisposing to, and existing in association with, adenocarcinoma of the colon and esophagus. This evidence suggests that many of the important events in the development of colonic and esophageal adenocarcinoma first occur in tissues that are neither highly dysplastic nor malignant.
Supported by UNESCO / MIRCEN network © 2000 by Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej00018f6.jpg] [ej00018f3.jpg] [ej00018f1.jpg] [ej00018f5.jpg] [ej00018f4.jpg] [ej00018f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}