 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
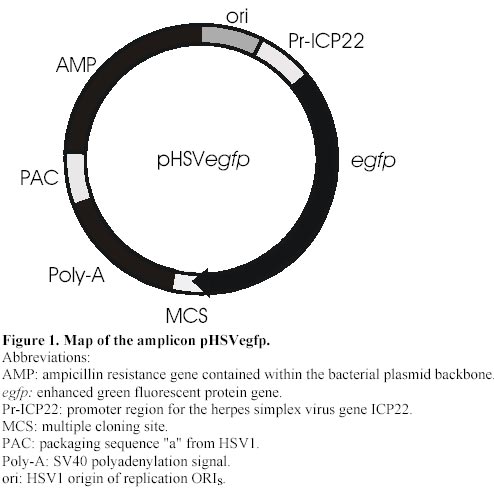
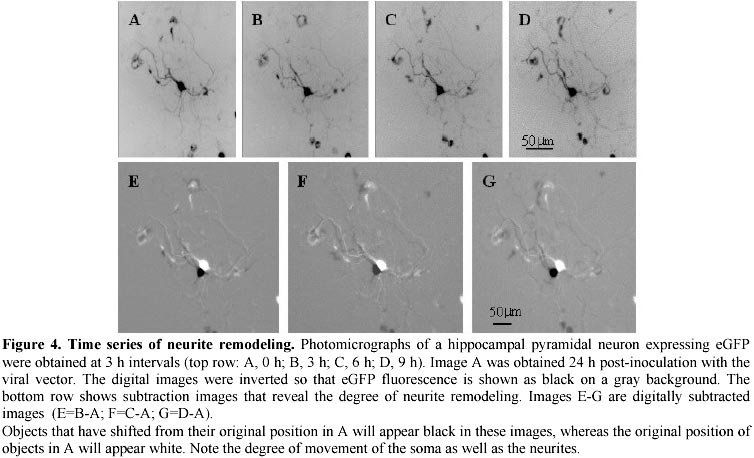
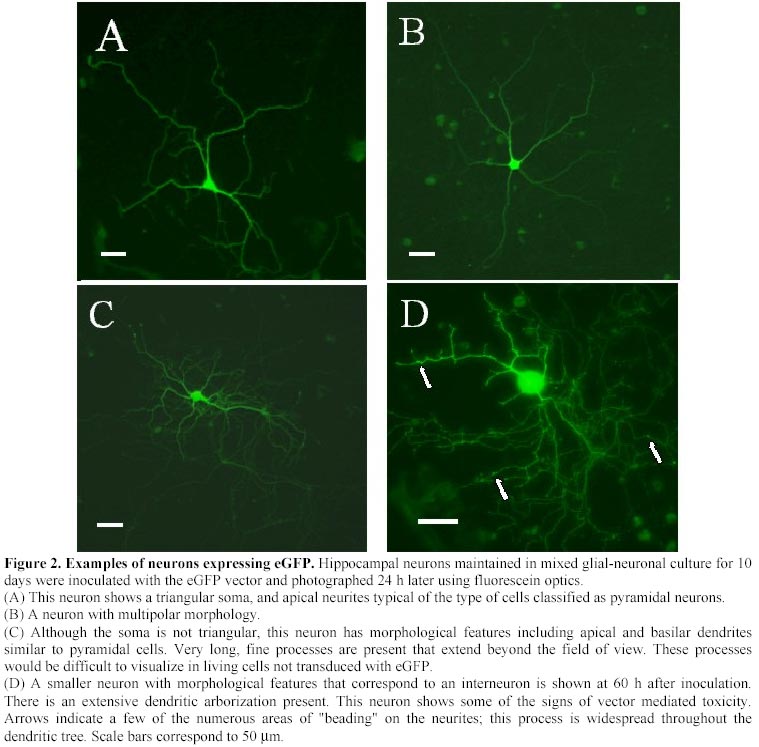
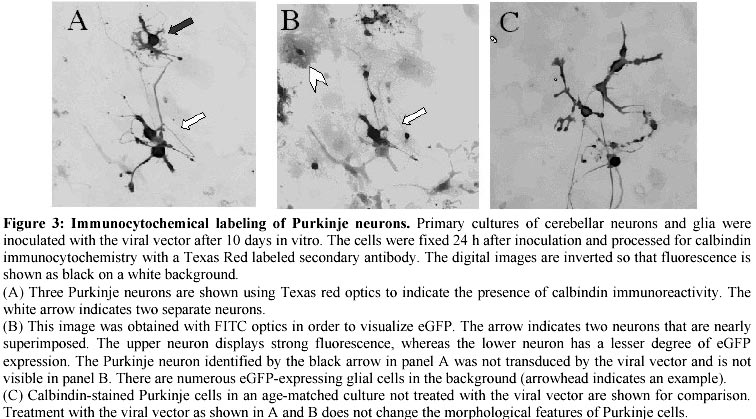
Electronic Journal of Biotechnology, Vol. 4, No. 1, April, 2001 A herpes simplex viral vector expressing green fluorescent protein can be used to visualize morphological changes in high-density neuronal culture Torsten Falk1, Lori A. Strazdas2, Rebecca S. Borders3, Ramsey K. Kilani4, Andrea J. Yool5 and Scott J. Sherman*6 1Department
of Physiology, The University of Arizona, PO Box 245051, 1609 N Warren Ave., Tucson,
AZ 85724, USA Phone: 520-626-2044 Fax: 520-626-2383 E-mail: Tfalk@u.arizona.edu
Financial support: NIH grants 5 K08 NS 02015-03 (S.J.S.) and 1 R01 MH59747-01A1 (A.J.Y.); Epilepsy Foundation of America Fellowship (S.J.S.); and Small Grants Program funding from The University of Arizona (S.J.S.). Received
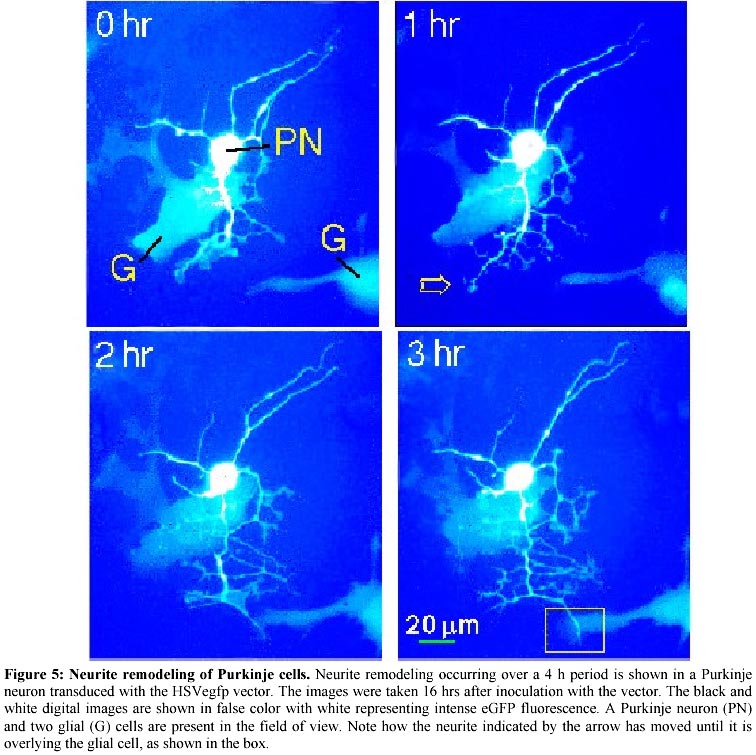
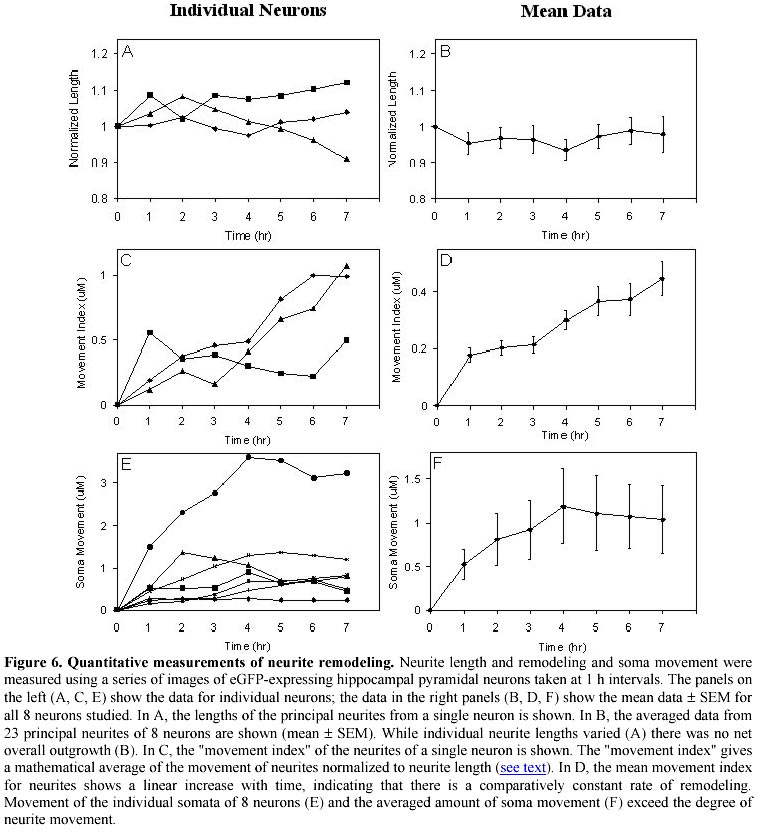
December 13, 2000 Code Number: ej01005 Abstract High-density cultures of mammalian neurons offer a model system for studies of brain development, but the morphological features of individual neurons is difficult to ascertain. We show that a herpes virus vector expressing a bioluminescent protein allows detailed morphometric analyses of living neurons in complex culture environments. Expression of enhanced green fluorescent protein (eGFP) was constitutively driven in neurons using the herpes simplex virus amplicon system. This system allowed us to make novel observations regarding development in high-density cultures from rat hippocampus and cerebellum. After the phase of initial neurite outgrowth, maturing neurons continue to show rapid remodeling of the neurite branches (0.79 ± 0.11 mm/h per neurite; mean ± SEM, n=8), and displacement of the soma within the neurite arbor (1.35 ± 0.74 mm/h). These results demonstrate that a substantial capacity for morphological plasticity persists in maturing mammalian CNS neurons after cessation of net neurite outgrowth in early development. Keywords: cerebellum, green fluorescent protein, hippocampus, plasticity, Purkinje neuron. Article Dispersed cultures of fetal neurons are commonly used as a model system to study brain development. When plated at higher densities that more closely approximate the connectivity found in vivo, the morphological features of individual neurons become indistinguishable among the dense tangle of the neuropil formation. Under these conditions, routine light microscopic analyses are not useful in studying morphological changes. We have employed a viral vector to induce expression of green fluorescent protein in dense heterogeneous cultures of neurons and glia. Using fluorescent microscopy, we demonstrate that detailed morphological analysis of single neurons in dense cultures is feasible. We have employed time-lapse photography to image the remodeling of neurites and somata of living neurons transfected with a virus engineered to express green fluorescent protein as an abundant cytoplasmic marker. Green fluorescent protein (GFP) is a bioluminescent molecule that has several advantages over other reporter genes: (1) its fluorescence does not depend on cofactors or substrates; (2) the protein is stable and not subject to rapid bleaching; (3) and visualization of living cells can be performed without obvious detriment to viability (Chalfie et al. 1994). Enhanced green fluorescent protein (eGFP) is a genetically modified, codon-optimized variant of the native protein found in the jellyfish Aequoria victoria (Zhang et al. 1996). Its fluorescence is 5 times brighter than that of the native protein when visualized with systems designed for fluorescein optics. GFP is finding great utility in the field of developmental neurobiology as a reporter gene and bioluminescent marker. The GFP gene has been incorporated in the genome of a variety of organisms during early embryonic development for the study of cell differentiation and for tracking patterns of neuronal migration (Jostock et al. 1998; Van den Pol and Ghosh, 1998; Okada et al. 1999; Spergel et al. 1999). Several studies have reported success in utilizing GFP in post-mitotic neurons using viral vectors (Aboody-Guterman et al. 1997; Smith et al. 1997; Gwag et al. 1998; Howe and McCarthy, 1998) and non-viral methods (Craven et al. 1999; Fernandez-Fernandez et al. 1999; Han et al. 1999; Watanabe et al. 1999; Eldadah et al. 2000). The amplicon-based herpes virus vectors (Breakefield and DeLuca, 1991) show promise for delivering genes into post-mitotic neurons and have been used to drive the expression of GFP in cultured striatal (Aboody-Guterman et al. 1997) and cortical (Coopersmith and Neve, 1999) neurons. We have extended these results, using the herpes amplicon system to transfer the eGFP gene into cultured hippocampal and cerebellar neurons. In this report we demonstrate that viral vector-mediated transgene expression of eGFP is useful for studying the dynamic morphological development of mammalian neurons maintained in primary culture. We are able to show morphological remodeling and soma movement in high-density hippocampal and cerebellar primary cultures. Materials and Methods Construction of pHSVegfp. The amplicon plasmid (pHSVegfp) was constructed from the plasmid pHSVlac (gift from Dr. A. Geller, Boston Children’s Hospital). The lacZ marker gene present in the original construct was excised from pHSVlac by endonuclease restriction cuts at the HindIII and EcoRI sites and a novel synthetic polylinker containing unique restriction sites was directionally cloned into the open site. This polylinker contained the sites: HindIII-AflII-BstXI-XhoI-NheI-BclI-ClaI-NsiI-DraIII-EcoRI. The eGFP gene was removed from a commercially-available construct (Clontech, Palo Alto, CA) at the HindIII and AflII sites and ligated into our polylinker. The resulting plasmid, pHSVegfp is illustrated in Figure 1. Packaging of the viral vector. The E5 cell line permissive for the propagation of the replication-deficient herpes virus was the gift of Dr. N.A. DeLuca (University of Pittsburgh). The E5 cell line was maintained in Dulbecco's modified eagle medium (DMEM, Gibco BRL) with 10% (v/v) fetal bovine serum. The E5 cells were transfected with the amplicon plasmid using Superfect cationic liposomes according to the manufacturer’s protocol (Qiagen). 24 h after the transfection, the E5 cells were then infected with the helper virus (d120HSV1 herpes virus, gift of Dr. N.A. DeLuca, University of Pittsburgh). The packaged virus was propagated using the E5 cell line following the protocol of Dyer and Tufaro (1997). Initial transfection and viral propagation were carried out when E5 cells were 70-80% confluent. E5 cells were lysed using a single freeze thaw cycle followed by sonication. E5 cells were harvested with a cell scraper, pelleted (1000 x g, 5 min), frozen in liquid N2, and thawed at 37ºC. Sonication was carried out using a water-cooled bath adapter and a Vibracell ultrasonic processor (Sonics and Materials, Newtown, CA) at 100% output for 2 min. Cell debris was removed by centrifugation (10,000 x g, 10 min), and the supernatant which contains the viral vector was stored in liquid N2. During the serial passage of the vector, the ratio of the helper virus to the packaged amplicon was monitored by a plaque assay using a lawn of E5 cells. The E5 cells were incubated with a serial dilution of the E5 lysate for 1 h and then washed with phosphate-buffered saline (PBS). A topping media was added that consisted of the growth media supplemented with 1% polyethylene glycol. Amplicon transformation was counted using fluorescein optics to tally the number of living eGFP-positive E5 cells at 24 h After 4 days, cells were fixed in 70% methanol and stained with 25% methylene blue in methanol. Plaques resulting from helper virus were counted under low power light microscopy after staining with methylene blue. Primary neuronal cultures. Timed-pregnant Sprague-Dawley rats (Harlan) were anesthetized by exposure to CO2 and sacrificed. The embryos (day E20) were removed, cooled on ice, and decapitated. The cerebellar cortices and hippocampi of embryos were dissected and gently minced and triturated in Ca2+ -free saline (136 mM NaCl; 5.3 mM KCl; 0.3 mM Na2PO4; 0.2 mM KH2PO4; 33.2 mM glucose and 43.8 mM sucrose; pH 7.4), then plated on 35 mm Petri dishes coated with poly-D-lysine (20 µg/ml, Sigma), in Minimum Essential Medium (MEM, Gibco BRL) with 10% (v/v) heat-inactivated horse and 10% (v/v) fetal bovine serum. The cultures were incubated at 37ºC in a humidified 5% CO2 incubator. From days 4-7 the cultures were treated with 0.02 mg/ml 5-fluorodeoxyuridine to inhibit the growth of mitotic cells. Subsequently, the cultures were maintained in MEM with 10% horse serum, changed every 3 to 4 days. For cerebellar cultures, the medium was supplemented with KCl (10 mM final concentration) in order to promote growth of Purkinje neurons. Imaging. Cultures were placed in a custom made water-jacketed holder attached to the stage of an inverted microscope and maintained at 37ºC. The studies were carried out in the normal serum-containing MEM culture medium. The bicarbonate-buffered pH of the growth medium was maintained by a constant stream of air with 5% CO2. Gray-scale images were taken with a cooled CCD camera attached to a Nikon Diaphot 200 inverted microscope under the control of PMIS-software package (Photometrics, Tucson, AZ) using a 500 ms exposure to UV light filtered with a FITC (B-2A) filter set. True color images were acquired digitally on a Nikon E800M microscope with a Hamamatsu C5180 camera, using Adobe Photoshop 5.0 software. Immunocytochemistry. Cell cultures were rinsed three times in MEM with 4% sucrose, pH 7.3 at 37°C, and were fixed in 4% paraformaldehyde (in MEM with 4% sucrose) for 30 min at room temperature. Cells were permeabilized by three 5 min washes with Stop/Permeabilizing buffer (S/PB): 0.05% Triton X-100, 0.1% bovine serum albumin in PBS, pH 7.3. Permeabilized cultures were incubated with 1 ml of primary antibody (3 h at room temperature), rinsed with S/PB (3 times, 5 min each), and then incubated with 1 ml of secondary antibody solution (1 h at room temperature). The primary antibody was anti-calbindin 28 k (monoclonal raised in mouse from Sigma Chemical Co. used at 1:400 dilution). The secondary antibody was Texas Red-labeled goat anti-rabbit IgG (Vector Laboratories Inc., Burlingame, CA) used at 1:250 dilution. Antibodies were diluted to the indicated concentrations in S/PB. After incubation with the secondary antibody, cultures were rinsed 3 times with PBS (pH 8.0, 5 min each) and visualized with fluorescein (B-2A) or Texas Red (G-2B) filters using a Nikon Diaphot 200 inverted microscope with 10x or 20x objectives (Plan 40, N.A. 0.55, equipped with correction ring for culture dish thickness compensation). Morphometric analysis. Subtraction images were generated with the PMIS-software. Analysis was done with NIH Image software using the macro "Neurite Labeling" (C. Thomas, Integrated Microscopy, University Wisconsin, 1994). Neurite lengths and soma area were measured by computer-assisted linking of individual neurite segments with a polygon tool. The Movement Index was determined as follows. A digital subtraction was performed on images of the neuron at different time points (Figure 4), such that the two images were, in effect, superimposed. For each principal neurite, the area between the superimposed images was determined using Scion Image software. The area was divided by average length of the neurite for the time period under study. This value was termed the Movement Index. Controls for specimen drift were carried using marked culture dishes. There was no movement of the culture dish under the experimental conditions used for the morphometric studies. Results Transgene expression of the eGFP marker Figure 1 shows a map of the amplicon plasmid, pHSVegfp. The plasmid was constructed by excising the lacZ gene from pHSVlac and replacing it with the coding sequence for eGFP. The pHSVegfp plasmid was packaged as a viral particle using the replication-deficient amplicon system (Breakefield and DeLuca, 1991). This method utilizes a helper virus to provide the genes necessary for the structural components of the viral vector. The helper virus is missing a crucial gene (ICP4) and can only replicate by complementation in a specially engineered host cell (E5 Vero cell line) that has been transduced with the ICP4 gene (DeLuca et al. 1985). This method leads to the production of a viral vector that consists of two populations of viral particles: (1) those containing multiple, concatemeric copies of the amplicon plasmid and (2) a new generation of helper virus. The helper virus may produce toxicity due to the transcription of other immediate-early genes that disrupt native cellular protein synthesis. As the vector is repeatedly passaged in the E5 cell line, competition occurs between the two viral populations. The eGFP reporter gene is useful in following this competitive process with repeated passages, since E5 cells harboring the eGFP amplicon will fluoresce brightly. After passaging 3-5 times, >80% of the E5 cells fluoresce, and the amplicon to helper ratio is generally in the range of 1:1 to 1:4 as determined by plaque assays. Lysates of E5 cells typically contained a vector titer of ~105-106 plaque forming units (pfu)/ml when tested on a lawn of fresh E5 cells. The viral vector drives robust expression of eGFP in a variety of cultured neurons and glial cells. When 25-50 ml of lysate containing ~1000 pfu was added to the cultures, 10 to 25 transduced neurons were usually observed in each 35 mm culture dish at 24 h. We did not find any obvious neural specificity of this vector since glial cells were transduced at nearly the same frequency as neurons. Given these constraints, we have found this system to be useful in transducing several individual neurons within each microscopic field and therefore suitable for the morphological or electrophysiological study of individual neurons. The viral transduction rate is not sufficiently high to carry out studies using homogenized samples from the entire dish, since the majority neurons are not transduced. The eGFP protein appears to distribute throughout the cytosol in a homogeneous fashion and penetrates fine neuritic processes. The morphological characteristics of living cultured neurons are easily determined as shown in Figure 2 depicting hippocampal neurons maintained in mixed neuronal/glial primary culture (10-14 days in vivo). These neurons were grown in high-density cultures and white-light or phase contrast optics disclosed up to 50 somata with a dense tangle of neuritic processes in the field of views shown. The transgene expression of eGFP is very useful for studying the morphology of individual neurons in these complex culture conditions. Even in cases where neurons were in sparsely covered portions of the culture dish, we found that eGFP allowed visualization of finer processes than were visible by phase contrast. Although not studied quantitatively, it was observed that neurons displayed the highest intensity of fluorescence at 24-30 h after addition of the vector to the culture. At 16 h, transduced neurons were visible, but the intensity and number of transduced cells increased over the next 8 hours. After 48 h, there was a slow decline in the number of transduced neurons. The possible reasons for this decline are discussed later in the section on Vector-Induced Cytotoxicity. We sought to test whether the vector-induced expression of eGFP could be carried out in sequence with antibody labeling, in order to identify transduced neurons of a specific subtype. There are no protein markers that can uniquely identify pyramidal neurons in dispersed hippocampal cultures; instead these neurons are commonly classified in vivo by their anatomic features and location within the laminar structure of the hippocampus. Thus, it is problematic to correlate morphologically identified hippocampal neurons in vitro with their in vivo counterparts. On the other hand, dispersed cultures of cerebellar neurons contain Purkinje cells that can be positively identified by the presence of calbindin, which is not found in the other cell types within the cerebellum (Wassef et al. 1985). Using cerebellar cultures, we tested whether double labeling with the eGFP marker and calbindin could be carried out. We labeled cerebellar cultures with anti-calbindin primary antibodies and a secondary antibody coupled to Texas Red, which fluoresces at a wavelength enabling differentiation from the eGFP signal. The cerebellar cultures (10 days in vitro) were inoculated with the pHSVegfp viral vector 24 h prior to processing for calbindin immunoreactivity. Figure 3 shows the results of one of these labeling experiments. The eGFP marker intensity was not diminished by fixation and thus transgene expression of eGFP can be coupled with a second immunochemical marker for use in double-labeling protocols. Time lapse studies Transgene expression of eGFP could be particularly useful in tracking morphological changes in living neurons. In order to establish the viability of this technique, eGFP-expressing neurons were studied over a time period of up to 10 h using time-lapse photography with fluorescein optics. Photographs were obtained every 45-60 min. Figure 4 (A, B, C, D) shows a series of time lapse images of an eGFP-expressing hippocampal neuron. The top row shows a series of images of a neuron collected over a 9 h period during which active remodeling of the neuritic processes and migration of the soma is evident. The first image in the series was obtained 20 h post-inoculation with the viral vector. In order to highlight these changes in morphology, the initial image A is digitally subtracted from the images obtained at 3, 6, and 9 h (Figure 4; A, B, C, D). White represents those parts of image A that are no longer fluorescent in images B, C, or D; and black represents those parts of images B, C, and D that were not fluorescent in image A. The subtracted images (Figure 4; E, F, G) show progressive changes in the position and shape of the neuron as compared to the starting position. Some of the change is due to movement of the soma and associated movement of the proximal portions of the attached neurites. Distal portions of the neurites also show divergence over time that appears to be independent of the soma movement. Remodeling of neurites was also found in cultured Purkinje cells (Figure 5). The changes are not due to specimen drift: (1) there was no movement of a marker placed on a culture dish and observed under comparable experimental conditions; (2) objects viewed in lower power fields show divergent movement; and (3) images obtained 1-2 min apart can be superimposed, ruling out sources of artifact due to more rapid oscillations in position. In order to further study neurite remodeling, quantitative morphometric measurements were carried out on time lapse series of eGFP-expressing hippocampal pyramidal neurons. The length of each primary neurite (not including secondary branches) was measured as a function of time, in order to determine whether there was consistent outgrowth (as seen in the first few days after plating) as opposed to neurite remodeling without a change in net length. We studied 23 of the principal neurites of 8 neurons for a period of 7 h. Only neurites that could be entirely contained in one field of view were studied. It is likely that most of the neurites studied were dendrites, since the long processes that are characteristic of axons typically extended beyond the range of the field. Figure 6 shows a compilation of this data. For purposes of comparison between cells, the neurite length measurements have been normalized to the measurement at time zero. In terms of absolute measurements, the length of the primary neurites (not including secondary branches) varied from to 2.6 -96 mm. Data from a single neuron is shown in Figure 6A. In this example, some neurites increased in length while others decreased over the 7 h time period. The averaged length of the neurites (41.1 ± 5.11 mm) did not significantly change (Figure 6B) over time, indicating that the morphological changes observed are not neurite outgrowth but instead represent active neurite remodeling. We also studied the lateral shift of the neurites across the surface of the growth substrate in a quantitative manner. Subtraction images such as shown in Figure 4 were used for morphological measurements. The difference in position between the initial image (white) and a subsequent image (black) was approximated with computer-assisted analyses using the polygon tool of an imaging program. Integrating the area between the white and black boundaries sums the amount of lateral movement over the full length of each neurite. The measurement was divided by the average neurite length over the time period studied in order to give a normalized value for neurite mobility. This value, termed the Movement Index, corresponds to the average lateral movement of the neurite. Figure 6C shows the lateral movement indices for the neurites of a single neuron and demonstrates that neurite remodeling occurs throughout the observation period. The averaged results from 23 neurites observed over an 8 h period are shown in Figure 6D. We also tracked movement of the somata using a similar method: data from 8 neurons are shown individually in Figure 6E; the averaged data are shown in Figure 6F. The mean movement of the somata exceeds the mean movement index of the neurites, suggesting that these processes are independent and perhaps controlled by separate regulatory pathways. Even when the outlying data from the somata showing the most extensive movement are excluded from the analysis, the initial rate of soma movement (1.35 ± 0.74 mm/h) still exceeds the rate of neurite movement (0.79 ± 0.11 mm/h). This finding suggests that the soma can migrate to different positions within a web of neurite branches. Vector-induced cytotoxicity As viral vector systems gain attention as tools for transgene expression, it is important to document the current limitations of this approach. When we attempted to carry out morphological measurements of neurons over longer time frames, we noted toxic effects that became evident in many neurons 3 to 4 days after inoculation with the vector. In contrast, untreated controls remain healthy for several weeks. These toxic effects of viral infection become lethal, as many fluorescent ghosts appeared in the culture after 4 to 5 days post-inoculation. We have observed two morphological signs after 48 h that precede cell death: (1) the soma becomes progressively more rounded and (2) bead-like formations occur on the neurites (Figure 2D). This process is not uniform; occasionally these signs of toxicity are seen within the first 24 h of transgene expression; in other cases, healthy-appearing neurons can be found, although in reduced numbers, as late as 4-5 days post-inoculation. Discussion Primary cultures of rat hippocampal and cerebellar neurons have proven to be useful and versatile models of neuronal development. The ease of experimental manipulation of cells maintained in vitro has allowed elucidation of several signal transduction cascades that are important in regulation of neurite outgrowth (Audersirk et al. 1997; Hindley et al. 1997; Mattson et al. 1988; Brann et al. 1999). These studies have largely been performed in relatively sparse cultures that lack the capacity for extensive synapse formation. After synaptic contact is made with other neurons, it is likely that other regulatory processes come into play (Cabell and Audesirk, 1993; Schilling et al. 1991). In this report, we describe a process of neurite remodeling separate from outgrowth, occurring at a later developmental stage. Most other studies of primary hippocampal neurite outgrowth have been performed at 2-5 days after plating; our studies were performed on neurons at 8-12 days after plating. Neurites of these older neurons continue to show substantial movement in the absence of changes in net length during this development stage, and there is continued movement of the soma as well. To our knowledge, these dynamic and rapid morphological processes have not previously been quantified in cultured mammalian neurons. At this stage, we can only speculate about the in vivo correlate of our observations. It seems likely, however, that the neurite and soma movements studied in this report reflect the same processes that give rise to neuronal migration and shaping of the dendritic arbor in the intact nervous system. It is of interest to note that the rate of soma movement exceeded the rate of movement of the neurites. This suggests that the neurites may provide anchoring sites for the neuron while allowing relatively more freedom for soma movement. This is reminiscent of the process occurring in vivo by which cell somata find their position within laminar structures of the brain such as the hippocampus and cortex. The methodology described here will be useful in elucidating the cellular mechanisms that underlie these phenomena. The fluorescence also facilitates electrophysiological study using patch clamp techniques since the morphology can be determined prior to cell selection and could eliminate the need for dye filling. The results of electrophysiological studies of eGFP-expressing neurons show no significant changes up to 48 h after inoculation with the viral vector (Falk et al. 2001). Vector-mediated transgene expression of eGFP has advantages over other methodologies. Despite recent advances (Kaech et al. 1996; Kohrmann et al. 1999), non-viral transformation of primary neurons remains difficult, and dye loading is considerably more laborious. The herpes amplicon can easily accommodate bi-cistronic constructs (Breakefield and DeLuca, 1991) and eGFP can be expressed as a fusion protein without altering the function of structural elements (Fischer et al. 1998; Craven et al. 1999), ion channels (Siegel and Isacoff, 1997), and other cellular components (Haubensak et al. 1998). Thus, antisense knockdown or over-expression of target genes (Jareb and Banker, 1998) can be carried out and the effect on morphological development easily monitored. Cytotoxicity remains a limitation of the viral vector technique described here. We believe that within the 24-48 h time frame described here, the helper-dependent system can be used with confidence. This time frame is consistent with the report of Jareb and Banker (1998), who also found delayed cytotoxicity when they utilized the herpes amplicon system to over-express proteins in cultured hippocampal neurons for studies of neuronal polarity. There are a number of possible reasons for this variable toxicity. In untreated primary cultures, there is always a progressive reduction in the number of neurons that begins after the first week in vitro and continues throughout the 5-7 week life span. So, for a proportion of the eGFP-expressing neurons, we may be observing a natural process of cell death. However, our observations indicate that as the dose of the viral vector increases, the signs of toxicity become more evident. This toxicity is present not only in the eGFP transduced neurons but in non-fluorescent neurons as well. This indicates there is a component of toxicity induced by the helper virus or other components of the E5 cell lysate. In our hands, this toxicity places a practical limit on the extent of transformation that can be carried out with this vector system. We have found the dose limit to be about 500 ml of E5 cell lysate (~2-5 x 105 pfu) which is sufficient to transform 250 to 500 neurons per 35 mm culture dish. Higher doses produce toxicity evident at 24 h post-inoculation. In cultures inoculated with a lower dose of vector, individual transduced neurons display toxicity after a 3-4 day latency. This occurs even when the estimated multiplicity of infection is less than one. Under these conditions it is unlikely that an individual neuron would be infected with both helper virus and an amplicon-containing viral particle, unless there is a process of viral particle aggregation present. Therefore, it is possible that toxicity can also result from high-expression of the eGFP transgene. Future studies will employ a second generation amplicon system that is completely free of helper virus (Fraefel et al. 1996). It is likely that the elimination of helper virus-induced toxicity will extend the 48 h time frame considerably. It will also allow an independent evaluation of toxicity due to transgene expression eGFP alone, which is important in light of the findings reported here, as well as those of a separate report that eGFP expression can promote apoptosis (Hsiao-Sheng et al. 1999). Studies utilizing transgenic animals, retroviral infection, and in vivo injection of viral vectors into neural tissue demonstrate that expression of eGFP is compatible with long term neuronal survival and argue against significant toxicity due to eGFP expression alone (Blomer et al. 1997; Peel et al. 1997; Klein et al. 1998). The level of eGFP expression may be important, however; very high levels of protein expression may exhaust cell resources or lead to toxicity by other mechanisms. We have demonstrated that the neurites of densely-plated cerebellar and hippocampal neurons in primary culture undergo an active process of remodeling in the absence of net neurite outgrowth. This observation was made possible by using a herpes virus amplicon-based vector to drive transgene expression of eGFP. Improvements in this methodology, including the use of the helper-virus-free vector system, inducible promoter elements, and cell-specific promoters should extend the window of observation by reducing vector-induced cytotoxicity. Bi-cistronic constructs will allow experimental manipulations of proteins involved in the developmental regulation of neurite remodeling and easy identification of transduced neurons. This methodology should aid in the exploration of processes occurring after the formation of a neural network in densely plated primary cultures. Acknowledgements We thank Dr. R. Sloviter for assistance with microscopy, and B. Peterson for technical assistance. References
Supported by UNESCO / MIRCEN network © 2001 by Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej01005f5.jpg] [ej01005f6.jpg] [ej01005f2.jpg] [ej01005f1.jpg] [ej01005f3.jpg] [ej01005f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}