 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Electronic Journal of Biotechnology, Vol. 5, No. 3, December, 2002 Genomic sequence derived simple sequence repeats markers: A case study with Medicago spp. Viswanathan
Mahalakshmi1, P. Aparna2,
S. Ramadevi3,
Rodomiro Ortiz * #4 1International
Crops Research Institute for the Semi-Arid Tropics (ICRISAT), Patancheru, 502
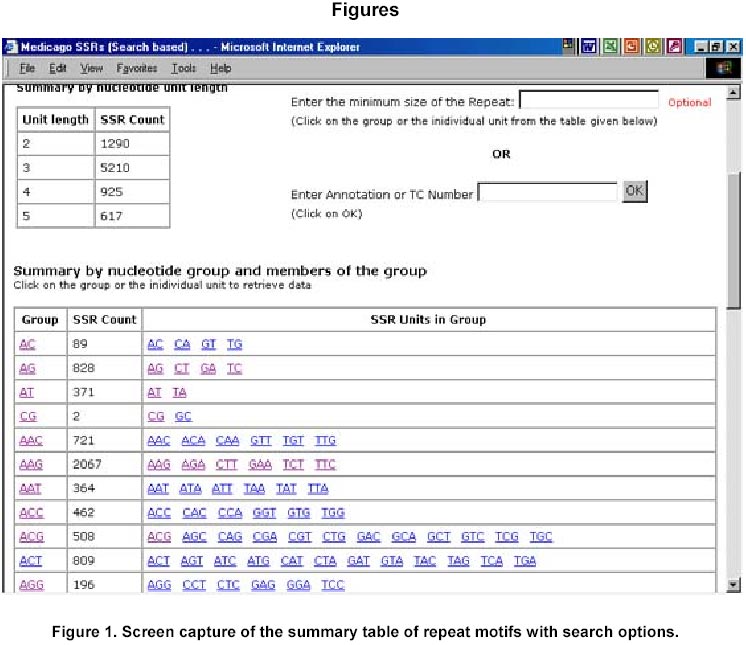
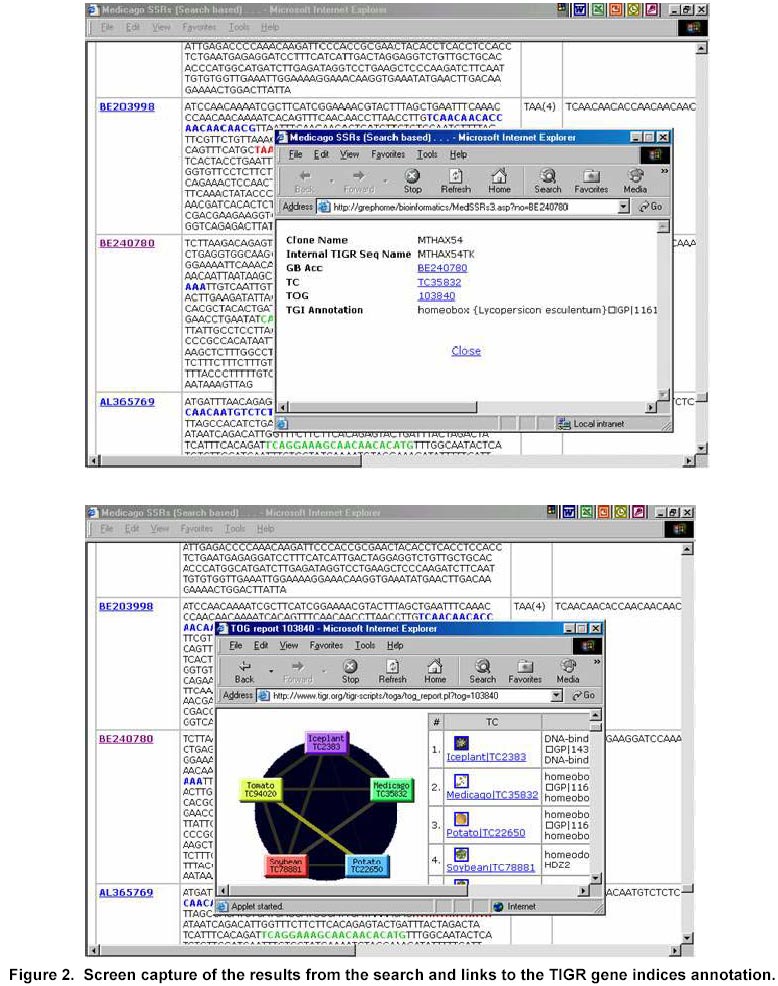
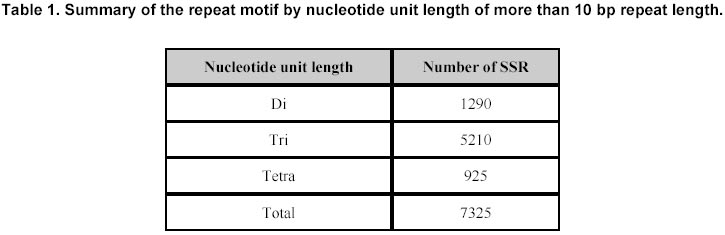
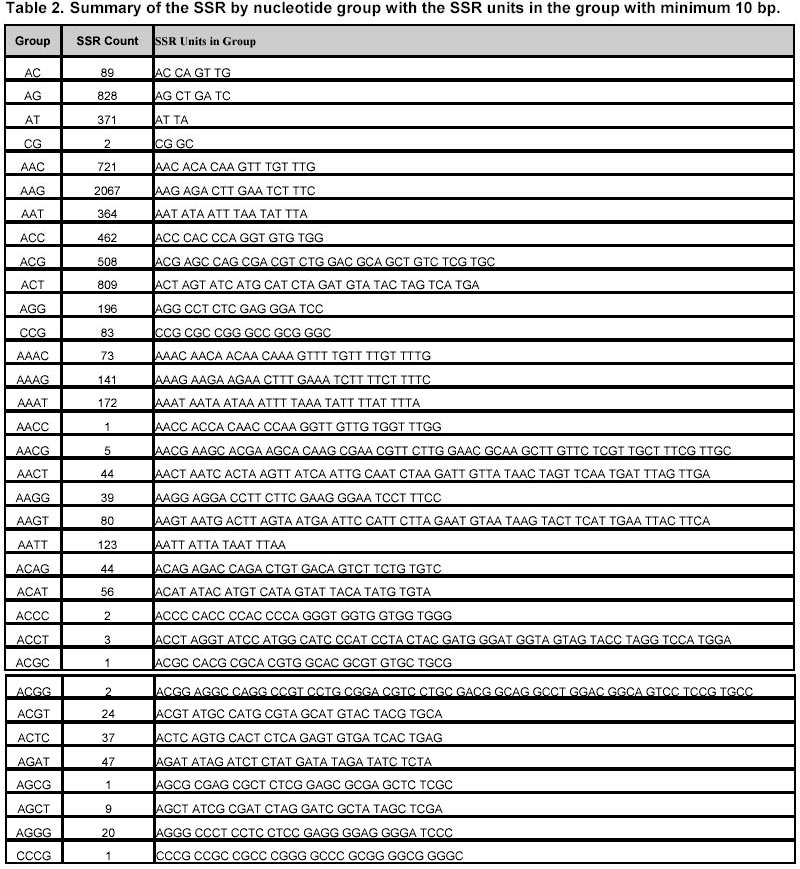
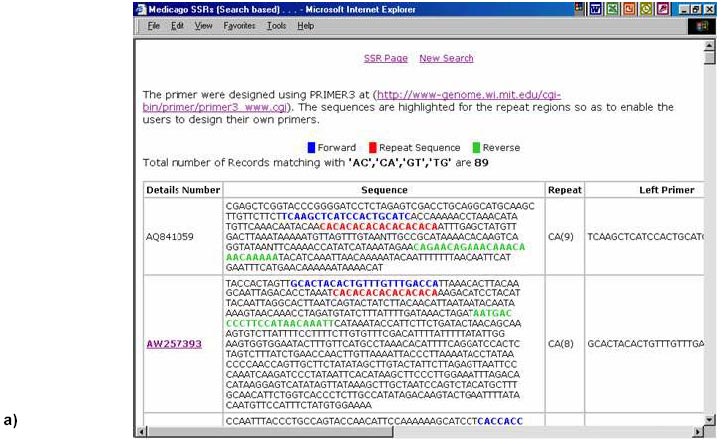
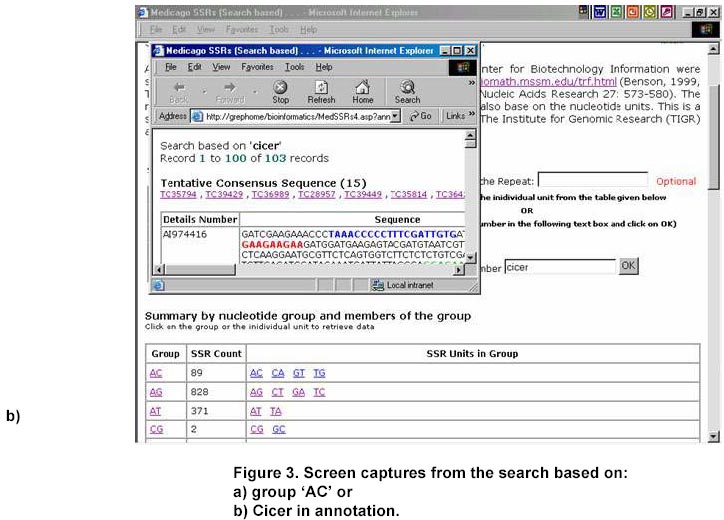
324, Andhra Pradesh, India, Tel: 91 40 3296161, Fax: 91 40 3241329 E-mail: v.mahalakshmi@cgiar.org Received May 14, 2002 / Code Number: ej02037 Abstract Simple sequence repeats (SSR) or micro-satellites are becoming standard DNA markers for plant genome analysis and are being used as markers in marker assisted breeding. De novo generation of micro-satellite markers through laboratory-based screening of SSR-enriched genomic libraries is highly time consuming and expensive. An alternative is to screen the public databases of related model species where abundant sequence data is already available. All the genomic sequences of Medicago from the public domain database were searched and analysed of di, tri, and tetra nucleotide repeats. Of the total of about 156,000 sequences which were searched, 7325 sequences were found to contain repeat motif and may yield SSR which will yield product sizes of around 200 bp. Of these the most abundantly found repeats were the tri-nucleotide (5210) group. Except for a very small proportion (436), these link to the gene annotation database at TIGR (http://www.tigr.org). To facilitate further exploration of this resource, a dynamic database with options to search and link to other resources is available at (http://www.icrisat.org/text/research/grep/homepage/genomics/medssrs1.asp) and on CDs from V.Mahalakshmi@cgiar.org. Keywords: bioinformatics, genetic markers, repeat motifs, simple sequence repeats, SSR. ArticleAmong various DNA duplication events, micro-satellites also referred as simple sequence repeats (SSR) are stretches of DNA containing tandem repeating di-, tri-, or tetra nucleotide units ubiquitously distributed throughout the eukaryotic genomes. They are found to be abundant in plant genomes and are thought to be the major source of genetic variation in quantitative traits. Simple sequence repeats originate from the unequal crossing-over or replication errors resulting in formation of unusual DNA secondary structures such as hairpins or slipped strands (Pearson and Sinden, 1998). If the resulting repeats happen to be in the coding region then it may be translated into single amino acid repeats or oligo-peptide repeats and can eventually dictate the structure of the protein and its function. SSR are becoming the standard DNA markers for plant genome analysis and are being used in markers in marker-assisted breeding. A wide variety of methods for construction of libraries enriched for micro-satellite sequences have been reported, the most popular among those being the ones based on vectorette PCR using anchored primers (Lench et al. 1996). The development of micro-satellite markers through these laboratory-based screening of SSR-enriched genomic libraries is highly time consuming and expensive. An alternative in well-studied species where abundant sequence data is already available is to use bioinformatics to screen these databases for sequences that contain SSR. Beyond the cost savings, this also offers the possibility of identifying STMS markers with rare SSR motifs for which it would be uneconomical to enrich through laboratory-based protocols. Finally, even for lesser-studied crops, this approach offers some potential for low cost development for limited number of markers, through the screening of related and allied crop. The level of polymorphism detected by such markers has yet to be fully tested. Nevertheless, markers developed in this way present a valuable resource for subsequent comparison between the model species and the related species. Arabidopsis thaliana, the first plant genome to be sequenced is considered the model crop for divots while rice (Oryza sativa) is the model crop for cereals. Recent availability of complete genome sequences has allowed analysis of SSR at whole genome level for many eukaryotic organisms including Arabidopis (http://www.ncl-india.org/ssr/index.html). Recently, Monsanto published about 6655 (http://www.rice-research.org/rice_ssr.html) segments of sequences with repeat motifs. These SSR and flanking DNA sequences were derived from the genomic sequences of the Monsanto rice genome project. Both these resources were developed using genomic sequence data (Barry, 2001). These SSR sequences included in these database include mono, di-, tri-, and tetra-nucleotide repeats of 24 bp or greater, and is expected to be used to expand knowledge of rice genetics and accelerate breeding research in rice and other crops around the world. International efforts to sequence, Medicago truncatula as the nodal species for comparative and functional legumegenomics is underway (http://www.medicago.org), which will pave way for the genetic improvement of the legume crops world wide. The molecular genetic map of the Medicago with agronomically important genes was recently published which should pave the way for comparative legume genomics (Thoquet et al. 2002). Though the molecular maps for legume crops of the semi-arid tropics (chickpea, groundnut and pigeon pea) are not very dense compared to the other related legume crops (i.e. soybean) but sequence data from Medicago and these related species could be mined to develop maps and markers. Today, we have the unique opportunity to share and link information from other resources around the world on the World Wide Web. Tools and resources are being developed to interpret the sequence in terms genes and their functions. The Institute for Genomic Research (TIGR) (http://www.tigr.org) has assembled the gene annotation or indices for various crops including rice (Yuan et al. 2001) and Medicago (http://www.tigr.org/tdb/mtgi) that are a set of non-redundant transcripts from the public Plant expressed Sequence Tag projects. This has become a valuable resource for researchers in crop and plant genome project. The research in this article provides information on searching publicly available genomic sequences of Medicago for repeat motifs, which might be used in further development of SSR for testing with other legume crops. Materials and Methods All the nucleotide sequences related to Medicago from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov) were downloaded in FASTA format and analysed for the repeat patterns using the tandem repeat finder program at http://c3.biomath.mssm.edu/trf.html (Benson, 1999). The gene indices database from TIGR was also downloaded. The local database containing the entire sequences in FASTA format, the repeat motif and the potential primer and the gene indices was created in a relational database (SQL7.0TM ). The resultant database of the repeat motifs was analysed to classify the patterns, their occurrence and abundance. The results of this analysis have been compiled in the form a database and are available at http://www.icrisat.org/text/research/grep/homepage/genomics/medssrs1.asp. A search facility with an option to select the size of the repeat motif (default 10 bp) and the pattern (either a group or unit in the group) or annotation is provided at this site (Figure 1). The result from the output of the SSR describes the accession number in the GenBank (http://www.ncbi.nlm.nih.gov), the sequence in FASTA format with repeat, potential flanking primer regions highlighted in different colours, the repeat motif with the number repeats, the left primer, the right primer, the annealing temperatures, the GC% of the primers and the product size. The accession number cross-linked to the TIGR gene annotation database by a hyper-link, which gives full information on the tentative consensus (TC) group of the TIGR database and gene annotations that are available. Full information, including the ortholog group (TORG) information, is displayed (Figure 2). Results For our study, we selected Medicago, which is considered the nodal crop for all legumes. All available (approximately 156,000) nucleotide sequences were analysed for the presence of tandem repeats up to 50 bp (maximum) length of repeat motif and no penalty gaps or indels were allowed. Although such methods may not detect some repeats containing insertions or deletions, it was considered that such a conservative approach might not be affecting the results very dramatically. The summary of the total number of potential SSR in each of the nucleotide length group is presented in Table 1. Of the total 7325, a little over 65% (5210) were from the tri-nucleotide groups. The di-nucleotide groups that potentially form the polypeptide repeats were the next largest group (Figure 1). The SSR summary by various nucleotide groups for a minimum repeat size length of 10 bp and the units that are part of the group, are presented in Table 2. The largest group is the tri-nucleotide group ‘AAG’ consisting of 2067 accessions. Of these, the largest sub-groups were from the ‘TCT’ and ‘TTC’ groups that code for serine and phenylalanine. In the di-nucleotide repeats the most common repeat was from the ‘AG’ group, which translated to di-oligopeptide of serine and leucine. Similar observations were reported by Katti et al. 2000 (http://www.ncl-india.org/trips) from the protein database though the found abundance of ploy-serine in longer chains. If the size of the motif is restricted to 18 bp and above, the number sequences is reduced from 2067 to 630. This flexibility allows the user to refine their choice for the repeat motif both in terms of pattern and size. The individual unit or group information provides the user with visual compiling of information on repeat region and possible primer regions. The search based on annotation or tentative consensus (TC) group displays all the sequences, which meet the annotation condition or TC number from the TIGR annotation database. Though number of sequences that meet the criteria for annotation, they may group into fewer tentative consensus groups, which is provided in the beginning on the results page. This information would facilitate limiting SSR from the same region (sequences form a TC) or can facilitate in retrieving information on SSR from a particular region. If the region or TC is associated with a function this would facilitate in development of multiple markers of which some may show polymorphism. An annotation-based search with the word Cicer (chickpea) retrieved 103 accessions (Figure 3 a, b) though the number of nucleotide accessions deposited in the Genbank for chickpea was 356 (as on 4/4/2002). These 103 accessions group into 15 tentative consensus groups. Discussion Data mining encompasses the use of pattern recognition technologies and statistical techniques to examine large amounts of data. De novo generation of micro-satellite markers through laboratory-based screening of SSR-enriched genomic libraries is highly time consuming and expensive. An alternative is to screen the public databases of related model species where abundant sequence data is already available. Beyond the cost savings, this approach also offers the possibility of identifying rare micro-satellite motifs, which would be uneconomical to identify through laboratory protocols. The availability of massive amounts of nucleotide sequence data has led to the development of innovative ways to examine these data as reflected in their functions. Data mining of public databases for marker development for crops not receiving sequencing attention was suggested as an approach (Mahalakshmi and Ortiz, 2001). A specialized database for all plant satellite repeats developed by Macas et al. 2002 and is available at http://w3lamc.umbr.cas.cz/plantsat. These are highly abundant tandem repeats and in contrast to micro- and mini-satellites their monomers are tens to thousands of nucleotides long and may not be very useful for developing micro-satellite markers for trait based crop improvement. Monsanto has made available to the public the data of genome sequence based 6655 SSR for rice from their rice genome initiative (http://www.rice-research.org; Barry, 2001) while for Arabidopsis thaliana, where the genome is fully sequenced, tandem repeats of nucleotides are available for II and IV linkage groups (http://www.ncl-india.org/ssr/ssr.htm). Since the advent of recombinant DNA technology in population genetics in the mid-1980s, the repertoire of genetic markers available for population studies and for crop improvement has increased enormously. Plant breeding has changed with the introduction of these molecular techniques. Molecular markers allow for the extension of traditional breeding methods with one important difference – to transfer greater variety of genetic information in a more precise and controlled manner. Marker-assisted selection for important but complex traits, which are often difficult to select in the routine breeding programs, will enhance the breeding programs in terms of better-focused products and save time and resources. Plant breeding or genetic improvement is based on the genetic variation within species that is usually expressed in terms of genetic alleles that occur at each of the given set of observable loci and the relative frequencies of their presence among individuals in the population. Polymorphism in the form DNA sequence variation within the transcribed regions of the gene may result in the phenotypic differences between individuals. Since the advent of molecular markers various types of DNA markers have been used in plant breeding and of these the most extensively used are the micro-satellite markers. The reasons for their extensive use are due to their mode of transmission, which is bi-parental-nuclear with few loci and many alleles per locus, mode of gene action being co-dominance with the exception of null alleles at some loci, show large variation within populations and are generally found in non-coding regions, which may contribute to the genome stability. Genome sequence and protein sequence information is publicly available for large- scale analysis from GenBank at (NCBI http://www.ncbi.nlm.nih.gov/) and European Molecular Biology Laboratory (EMBL http://www1.embl-heidelberg.de/). Today, the search for a gene of interest starts with sequence information, including expressed sequence tags (EST). Genome related public databases are an invaluable part of the scientific community and most notably the model organism databases, have two major consumers: the focused scientific community actively studying that system, and the large scientific community interested in relating this specialized information to and from other systems. The thrust of any high-throughput facility is the creation of large, well-organized, rigorous datasets. The model system databases can be mined by other but related crop specialists to design markers for marker-assisted selection and –aided introgression methods. Such an approach can save valuable resources both in terms of time and funds. The white paper presented at the workshop in 2001 for U.S. Legume Crops Genomics suggested similar approaches for the legumes currently not receiving sequencing attention. (http://macgrant.agron.iastate.edu/Legume_Initiative/LegGenomicsPaper10Oct01.html). The paper suggests to take advantage of DNA marker technology, to develop a core set of at least 1000 sequence tagged sites or STS that are universal among all legume species might be developed. For legume crops with large chromosome numbers and low levels of DNA polymorphism, more than 1000 STS will be needed to provide meaningful comparative maps. These STS markers will begin with strategically chosen PCR-based markers that have already been developed, especially in pea, soybean, and barrel medic. Eventually, the STS core set will grow by mining legume sequence data in order to find highly conserved sequences shared by all legumes. The legume STS core set will immediately provide powerful tools for trait mapping and marker-assisted breeding in all legume species, including those with few marker resources available today. The STS core will also interconnect the genetic maps of different species, revealing cases where genome organization is highly conserved and where rearrangements have occurred. The STS core will also simplify the important task of fingerprinting germplasm collections and analysing molecular evolution within the legume family. Further the need to curate a centralized database be able to access the species-centric databases, acquire and access the required data interactively, and generate reports in a user-friendly, web-accessible, and graphical manner was also suggested. Such an integrated resource will benefit all legume species by making genetic and genomic information developed in a wide range of species available to all other species researchers via a web-based interface. An integrated database provides a platform for integrated data analyses. Such a merged database would facilitate pan-legume data mining. This would provide a synergistic utilization of the shared data. Our study is one in such effort in this direction. In our research, we found that about 5% of all the available genomic sequences of Medicago truncatula showed tandem nucleotide repeat of various length and patterns. Of these, the most abundant are the di and tri-nucleotide repeats. The analysis of type and frequency of SSR in plant genomes showed abundance of SSR in genomic DNA, decreased in the ESTs (Cardle et al. 2000). Similar patterns are reported in the rice genome sequence data of Monsanto (Barry, 2001) and for Arabidopsis thaliana (http://www.ncl-india.org/ssr/ssr.htm). The most frequently encountered di-nucleotide repeat from this databases were from the AG though the AT group also had fairly large number. Similarly the most abundant tri-nucleotide repeat group is the AAG. Though frequently repeated sequence of di and tri nucleotide were from the AT and ATT group in the rice database but in Arabidopsis database the most frequent in tri-nucleotide group were from the TCT group. It is possible that, the estimates and patterns observed here are skewed by the preponderance of coding region as most of the sequences are from the cDNA library while in the other crop species they come from the genome. The pattern and number of SSR motifs from the genomic and EST sequences were quite different with the predominance of AT in the genomic sequence while AG was predominant in the EST sequence in Arabidopsis (Cardle et al. 2000). The other obvious difference which can be attributed to these differences is the nature of the crop species and the family they belong to; i.e. rice being a poaceae and Arabidopsis being a brassicaceae while Medicago being a papilionoideae. The provision to link to the gene annotation data of TIGR gives additional information resource on the possible related functions. Searches based on gene annotation can also give the tentative consensus groups. In the absence of the information on the complete genome such information would help users to make the best bet options especially when the targets are function related. Though it is generally considered that SSR are found in abundance in the non-coding region of the genome, increasingly there is evidence that repeat motifs are found in EST and in proteins where motifs of single amino acids and poly-peptides are not infrequent (Katti et al. 2000). A total of 57.8 Mb publicly available rice (Oryza sativa L.) DNA sequence was searched to determine the frequency and distribution of SSR in the genome and a set of 200 SSR markers was developed and integrated into the existing microsatellite map of rice (Temnykh et al. 2001). Though these approaches are proving to be useful as SSR markers for the same species or closely related species within the same genus (SSR developed based on the information from Monsanto are proving polymorphic in rice and in Oryza sativa x Oryza glaberrima crosses (Dr. Marie-Noëlle Ndjiondjop, personal communication), their utility in other related species is yet to be tested. In maize such an approach using a very small number (280) of sequences yielded six SSR markers, which showed polymorphyism in eight maize inbreds and four of these co-segregated with genes from where they were derived (Senior and Heun, 1993). The strong synteny between related species would suggest that such approaches can be employed. For legumes, such as chickpea, pigeonpea, cowpea and groundnut, in which the availability of genomic sequences and DNA markers are limited, venturing into such approaches may provide better alternatives than to wait for the genomic information. The next logical follow up to this would be to test a set of randomly selected markers from this on Medicago trucatula (diploid), Medicago sativa (tetraploid), and other related legumes such as chickpea. The 14-consensus groups, which annotate to functions or proteins from Cicer can be the starting point for testing these SSR in Cicer (chickpea). Database availability The results from this study have been compiled in the form of a database and are made available at http://www.icrisat.org/text/research/grep/homepage/genomics/medssrs1.asp. The summary table provides a comprehensive information of the various repeats motif groups found in Medicago truncatula. Further the accessions are hyper linked to the TIGR database. If gene indices information is available from TIGR, links are provided to that database. This database is available on CD with the search and link facilities. Please write to V.Mahalakshmi@cgiar.org to get a copy of the same. Acknowledgments The authors would like to thank Ms B. Rekha, Mr. S. Prasad for meticulously searching and compiling the database and Ms. Y. Leela Devi for the programming support. References
Note: Electronic Journal of Biotechnology is not responsible if on-line references cited on manuscripts are not available any more after the date of publication. Supported by UNESCO / MIRCEN network © 2002 by Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej02037f2.jpg] [ej02037t1.jpg] [ej02037f1.jpg] [ej02037t2.jpg] [ej02037f3b.jpg] [ej02037f3a.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}