 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Electronic Journal of Biotechnology, Vol. 9, No. 5, October, 2006, pg. 604-609 TECHNICAL NOTE Deletion of DNA sequences of using a polymerase chain reaction based approach Pablo Pérez-Pinera1 , Manuel Menéndez-González*2, José Antonio Vega3 1Departamento de Morfología y Biología Celular,
Facultad de Medicina,
C/. Julián Clavería s/n, CP:33006,
Received September 22, 2005 / Accepted May 5, 2006 Code Number: ej06082 Abstract We developed a simple, rapid and reliable method to delete DNA fragments in plasmids using a polymerase chain reaction based amplification of the circular DNA sequence that excludes the fragment to be deleted. The primers are designed to contain a non-complementary 5' sequence consisting of a restriction enzyme target sequence. Following PCR amplification, the plasmid is digested with Dpn I to eliminate the template DNA, with the chosen restriction enzyme, and ligated. The only limitation is the selection of the restriction enzyme target sequence that must not be present in the original plasmid. The method is straightforward in its execution and success relies on a meticulous primer design that permits us obtain 100% of transformants containing the desired mutation. The extraordinary simplicity of the method makes it a valuable tool to generate DNA deletions in plasmids and to study the effects of those deletions in protein function. Keywords: deletion, mutagenesis, polymerase chain reaction, plasmid. Site-directed mutagenesis is a powerful method commonly used in many areas of molecular biology and biochemistry to study the effect of point mutations in protein activity or to study the role of specific domains in protein function. The first mutagenesis methods had very low rate of success and were not cost effective (Kramer et al. 1984). The description of polymerase chain reaction (PCR) (Saiki et al. 1985) turned mutagenesis much simpler as many ingenious methods were developed, most of them based on introduction of mismatches in the primers to generate mutations (Ling and Robinson 1995; Lanio and Jeltsch, 1998; Shenoy and Visweswariah, 2003; Chiu et al. 2004; Nagy et al. 2004; Rabhi et al. 2004; Wei et al. 2004; Ailenberg et al. 2005; Gaytan et al. 2005; Wu et al. 2005), although the look for clones with the desired mutations could be time consuming and not very efficient. The most common approach to delete DNA sequences in plasmids uses naturally occurring restriction sites followed by ligation with the plasmid (Allemandou et al. 2003). All those approaches require many different reactions, are not very efficient or reliable, and require repetition of the procedure many times before it successes. We describe in this manuscript a rapid PCR-based technique that takes advantage of the circularity of plasmid DNA to amplify the entire plasmid except for the region that is to be deleted. The deleted DNA sequence is replaced by the cutting sequence of a restriction enzyme plus the additional bases needed to keep the reading frame necessary to translate the DNA to protein. This restriction enzyme cutting sequence is introduced as non-complementary, non-overlapping strands in the 5' end of each primer. After PCR, a linear DNA product is generated whose 5' and 3' ends contain the sequence of the restriction enzyme chosen and, after digestion and ligation, the DNA fragment is recircularized and used to transform cells. Performing a Dpn I digestion step ensures that all the transformants contain a mutated plasmid and not source DNA. To illustrate this method we generated internal, N- and C-terminal deletions in the growth factor NT-3 previously cloned in the plasmid pcDNA6.2/GW/V5/D-TOPO. The new method requires only one PCR reaction, two digestions, a ligation and two purification steps therefore reducing the time to generate the mutants to one day and the efficiency observed was very high as all the transformants contained the desired mutation, kept the reading frame, were successfully expressed and no point mutations were observed. This technique is therefore a very valuable tool to delete different domains in proteins. A diagram of the steps followed is shown in Figure 1. The plasmid pcDNA6.2/V5/GW/D-TOPO, obtained from Invitrogen, Carlsbad, CA, with the sequence of neurotrophin 3 (NT-3) (GenBank Accession Number AY707640) was extracted from the E. coli strain JM109 and purified using the QIAprep Spin Miniprep Kit following manufacturer's recommendations. This method was used to construct three synthetic NT-3 derived proteins lacking the N-terminal region that we designated NT-3-N, an internal region designated NT-3-I and the C-terminal region designated NT-3-C. Primers were constructed to amplify the entire sequence of the plasmid except for the specific region that is to be deleted. The 5' ends of the primers are facing each other and 3' ends oriented so the extension will amplify the entire plasmid. The 5' ends of the primers include the cutting sequence of the restriction enzyme EcoRI (highlighted bold) with one adjacent base at the 5' and 3' ends to facilitate the digestion close to the end of DNA regions. As this sequence contains 8 bases, it is necessary to include an additional base in one of the primers to keep the polymerase reading frame between the restriction enzyme cutting sequence and the rest of the primer. The binding of the primers to the plasmid is shown in Figure 2. The sequences of the primers synthesized were NT-3 N-terminal deletion (NT-3-N): 5'-GGAATTCCAGCCAAGTCAGCATTCCAGCC-3', 5'-GGAATTCCCATGGTGAAGGGGGCGG-3'; NT-3 Internal deletion (NT-3-I): 5'-GGAATTCCAGAAGCCAGGCCGGTCAAAA-3', 5'-GGAATTCCCGGCTGGAATGCTGACTTGG-3'; NT-3 C-terminal deletion (NT-3-C): 5'-GGAATTCCAGCCGACCCAGCTTTCTTGTA-3', 5'-GGAATTCCACCGTTTTTGACCGGCCTG-3'. PCR was performed using 1 U of Platinum® Pfx DNA polymerase in a reaction containing 2 pg of the plasmid to be mutated, 1X Pfx buffer,
Gel extraction and purification DNA was purified from 0.9% agarose gels using the QIAquick Gel Extraction Kit, following manufacturer's instructions. Dpn I digestion, EcoRI digestion and ligation The gel-extracted PCR product was digested with 5 U of Dpn I and simultaneously with 1 U of EcoRI in a buffer containing
Plasmids were purified using the QIAprep Spin Miniprep Kit following manufacturer's instructions. Transfections and analysis of expression The plasmid pcDNA6.2/V5/GW/D-TOPO containing the sequence of NT-3 or the different mutants generated were transfected into 80% confluent HEK-293 cells growing for 2 days in EMEM supplemented with 10% Fetal Bovine Serum and 1% Penicillin/Streptomycin that was changed for OPTI-MEM-I 60 minutes prior to transfection. We prepared a mixture of 100 µl of OPTI-MEM-I, 6 µl of FUGENE 6 transfection reagent and 1 µg of the plasmid to be transfected. After 24 hrs HEK-293 cell lysates were prepared by incubating the cells in culture plates with 200 µl of a buffer containing
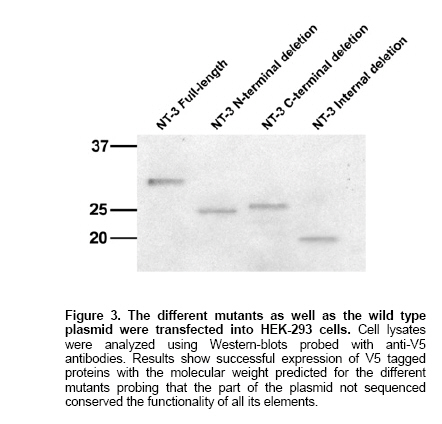
To test the method we used the vector pcDNA6.2/V5/GW/D-TOPO containing the sequence of human neurotrophin-3 (NT-3), previously cloned (GenBank Accession Number AY707640). We designed primers for PCR amplification as described in Methods and performed a PCR reaction using the template and the primer pairs described to generate N-terminal mutation but containing the starting codon, a C-terminal deletion but containing the V5 tag and an internal deletion. The sizes of the PCR products were the expected. The steps followed after the amplification are summarized in Figure 1. It is important to mention that the omission of the gel purification steps always resulted in unsuccessful transformations. We sequenced 10 transformants from every reaction and amplified using PCR with the first set of primers and another 40 to check the presence of insert with the correct size. All the samples sequenced contained the expected mutations, conserved the reading frame and no mutations were observed in the inserts, although the rest of the plasmid was not sequenced. The samples amplified using PCR contained inserts with expected size. We next tested whether the function of the rest of elements in the plasmid was preserved after the PCR performing transfections with the plasmids encoding NT-3 mutants and NT-3 full-length in cells HEK-293 and analyzed using Western-blot, the presence of the V5 tagged proteins using anti-V5 antibodies. The full-length NT-3 has a molecular weight ~31 kDa. The mutant NT3-N was designed to delete 228 base pairs near the 5' of the NT-3 gene. The resulting protein is expected to be ~25 kDa. The mutant NT3-I was designed to delete 336 base pairs in the middle of the NT-3 gene. The resulting protein is expected to be ~18 kDa. The mutant NT3-C was designed to delete 171 base pairs at the 5' of the NT-3 gene. The resulting protein is expected to be ~25 kDa. The results, shown in Figure 3 demonstrate similar levels of expression and expected size in the mutants what suggest that no critical changes have happened in the amplified plasmid. The sequences of the mutants generated are available in GenBank with accession numbers NT-3 N: AY841890; NT-3-C: AY841888 and NT-3-I: AY841889. In this paper we describe a method to delete DNA sequences in plasmids of any length using a PCR strategy that amplifies the entire plasmid except the sequence to be deleted. The only limitations are the efficiency of the polymerase used to accurately copy the DNA, what can be very high using proof-reading high fidelity polymerases, and the selection of a restriction enzyme that do not previously exists in the plasmid, what given the number of restriction enzymes commercially available is feasible. The key step is the proper selection of the PCR primers. The primer forward is designed to bind at the 3' region of the sequence to be deleted and is the sequence of the plasmid at that region containing in the 5' end the cutting sequence of a restriction enzyme not present in the rest of the plasmid. The primer reverse is the complementary and reverse of the plasmid sequence in the 5' end of the region to be deleted and contains in the 5' end the same sequence as the primer forward to be cut by a restriction enzyme, preceded by one or two bases to keep the reading frame and to avoid introducing one stop codon. It may also be important to consider the properties of the amino acids that are going to be introduced to avoid additional conformational or functional changes. The following step is to set a standard PCR reaction where the amount of template used should be kept as low as possible to minimize the contamination of the PCR product with wild type DNA. In our experience the use of 2 pg of DNA is optimal. The use of a proof-reading high fidelity polymerase is highly recommended, especially if the size of the plasmid exceeds 3 kb, to avoid undesired point mutations. The PCR product should be visualized using agarose electrophoresis and subsequently gel purified. Omission of this step results in our experience in very low number or absence of transformants probably due to decreased efficiency of the ligase in the presence of primers. Dpn I digests the adenomethylated DNA sequence 5'-GATC-3' (Lacks, 1980). This sequence is contained 25 times within the vector. The product of the PCR reaction includes the template methylated plasmid and DNA sequences amplified by the DNA polymerase that are not methylated; Dpn I eliminates template DNA and allows that in the subsequent transformation reaction only clones containing the DNA that was copied using PCR and carrying the desired deletion will be amplified. For this system to work the original plasmid has to be prepared in strains that have an intact dam methylation system. Most E. coli strains, like JM109, have an intact dam methylation system; however strains like JM110 are dam-deficient. The digested DNA is visualized using agarose electrophoresis and gel purified. Again the omission of this step has resulted in poor results. The ligated products are transformed into E. coli and purified. The reproducibility of this method depends on the primer design to perform the PCR reaction as the other steps are straightforward. The proper selection of a restriction enzyme whose cutting sequence is not present neither in the plasmid nor the insert, and that is able to easily cut DNA close to 3' and 5' ends on one hand and the selection of the optimal annealing temperature to perform the PCR reaction on the other hand are critical. If the PCR is successful and all the other instructions are carefully followed it can be expected to obtain 100% of transformants containing the desired mutation. The enzyme we chose to illustrate the method, EcoRI, is very efficient in cutting at 3' and 5' ends and we recommend its use when possible. Other enzymes that gave satisfactory results cutting close to the end of DNA fragments were KpnI or NheI. A complete list of cutting efficiency when close to the end for different enzymes can be found at the New England Biolabs Website: http://www.neb.com/nebecomm/tech_reference/restriction_enzymes/cleavage_linearized_vector.asp The use of non overlapping sequences at the 5' end of the primers may require decreasing the annealing temperature to setup the PCR reaction. However in our experience the optimal annealing temperature calculated using the program MacVector for a primer pair excluding the non-overlapping sequences permits proper binding of the primers and obtaining the desired PCR product. The advantages of this method are its high efficiency, very low cost and rapid generation of results. The efficiency is given by proof-reading polymerases that minimize the rate of miss incorporation of bases while copying DNA. Higher efficiency can be obtained if needed by increasing the amount of template and/or reducing the number of cycles. We have amplified successfully plasmids up to 7.5 Kb although, theoretically, the only limitation for longer plasmids would be the efficiency of the polymerase and presence of restriction sites. The low cost is given by the simplicity of the method that do not required specialized equipment, just a PCR thermocycler, or expensive reagents, just 1 U of polymerase, 5 U of Dpn I, 5 U of T4 ligase, and cells for transformation are required per reaction, what can be found in most molecular biology laboratories. The rapid generation of results as well as low cost are ensured by the high efficiency of the method and permits that one person during one working day can easily generate 12 mutations in one single protein with the warranty that, if all the steps are properly followed, the results will be completely satisfactory, avoiding long and time consuming protocols, expensive procedures, and saving the time and money needed to study a high number of transformants to find the ones containing the desired mutation as well as the frustration of repetitive negative results.
Note: Electronic Journal of Biotechnology is not responsible if on-line references cited on manuscripts are not available any more after the date of publication. © 2006 by Pontificia Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej06082f3.jpg] [ej06082f1.jpg] [ej06082f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}