 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Electronic Journal of Biotechnology, Vol. 11, No. 1, January 15, 2008 Diversity of bacterial communities in acid mine drainage from the Shen-bu copper mine, Gansu province, China Yu Yang#1, Wuyang Shi2 , Minxi Wan3 , Yanfei Zhang4 , Lihong Zou5 , Jufang Huang6 , Guanzhou Qiu#7, Xueduan Liu*#8 1School of Minerals Processing and
Bioengineering,
Central South University,
Changsha 410083, Hunan, China,
Tel: 86 0731 8877216,
Fax: 86 0731 8710804
E-mail: csyangyu@gmail.com Financial support: This work was supported by Chinese Science Foundation for Distinguished Group (No.50621063) and National Basic Research Program (973 Program) of P.R.China (No. 2004CB619201). Present address: #Key Laboratory of Biometallurgy, Ministry of Education, Changsha 410083, Hunan, China. Received January 19, 2007 / Accepted July 9, 2007 Code Number: ej08001 Abstract This study presents bacterial population analyses of microbial communities inhabiting three sites of acid mine drainage (AMD) in the Shen-bu copper mine, Gansu Province, China. These sites were located next to acid-leached chalcopyrite slagheaps that had been abandoned since 1995. The pH values of these samples with high concentrations of metals ranged from 2.0 to 3.5. Amplified ribosomal DNA restriction analysis (ARDRA) was used to characterize the bacterial population by amplifying the 16S rRNA gene of microorganisms. A total of 39 operational taxonomic units (OTUs) were obtained from the three samples and sequenced from 384 clones. Sequence data and phylogenetic analyses showed that two dominant clones (JYC-1B, JYC-1D) in sample JYC-1 represented 69.5% of the total clones affiliated with Acidithiobacillus ferrooxidans (γ-Proteobacteria), and the most dominant clones of JYC-2 and JYC-3 were affiliated with Caulobacter crescentus (α-Protebacteria). At the level of bacterial divisions, differences in the relative incidence of particular phylogenetic groups among the three samples and discrepancies in physicochemical characteristics suggested that the physico-chemical characteristics had an influence on phylogenetic diversity. Furthermore, the relationships between the discrepancies of physicochemical characteristics and the diversity of the bacteria communities in the three samples suggested that the biogeochemical properties, pH and concentration of soluble metal, could be key factors in controlling the structure of the bacterial population. Keywords: acid mine drainage, amplified ribosomal DNA restriction analysis, microbial communities.
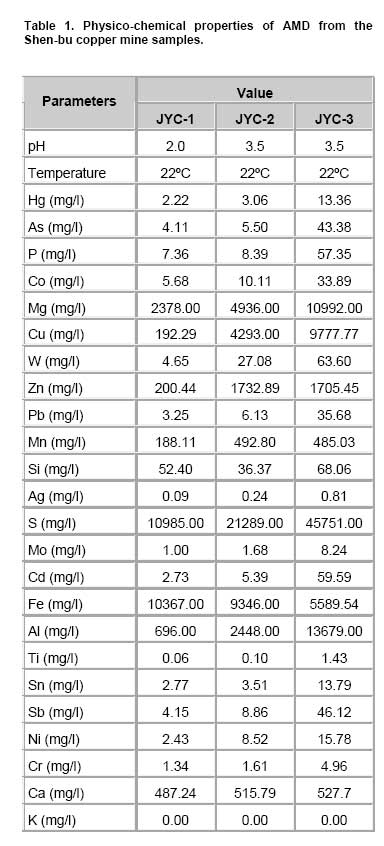
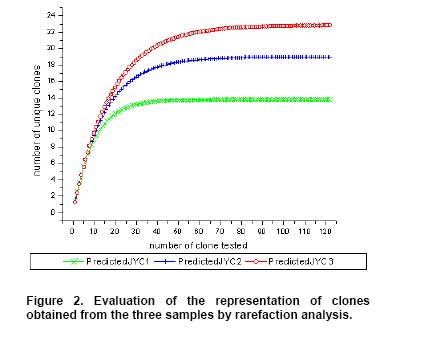
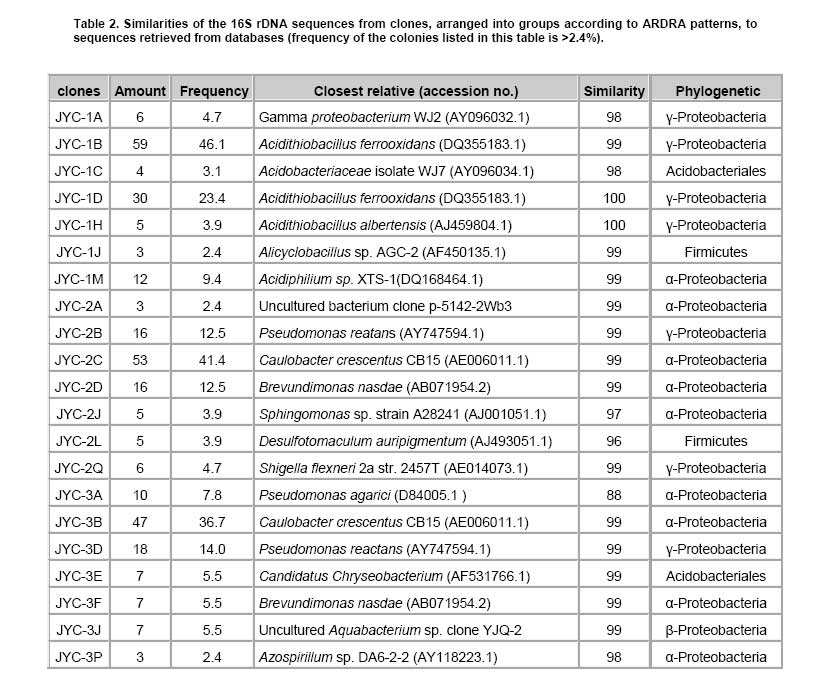
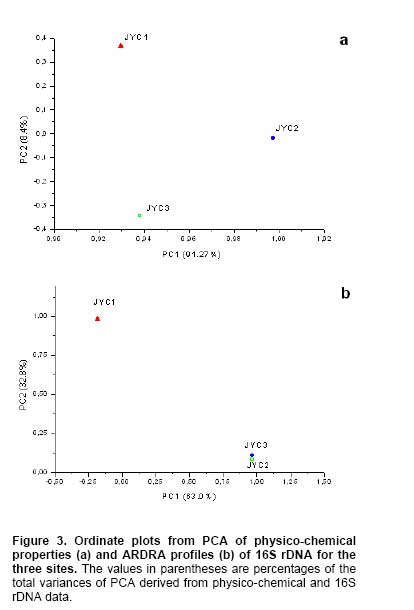
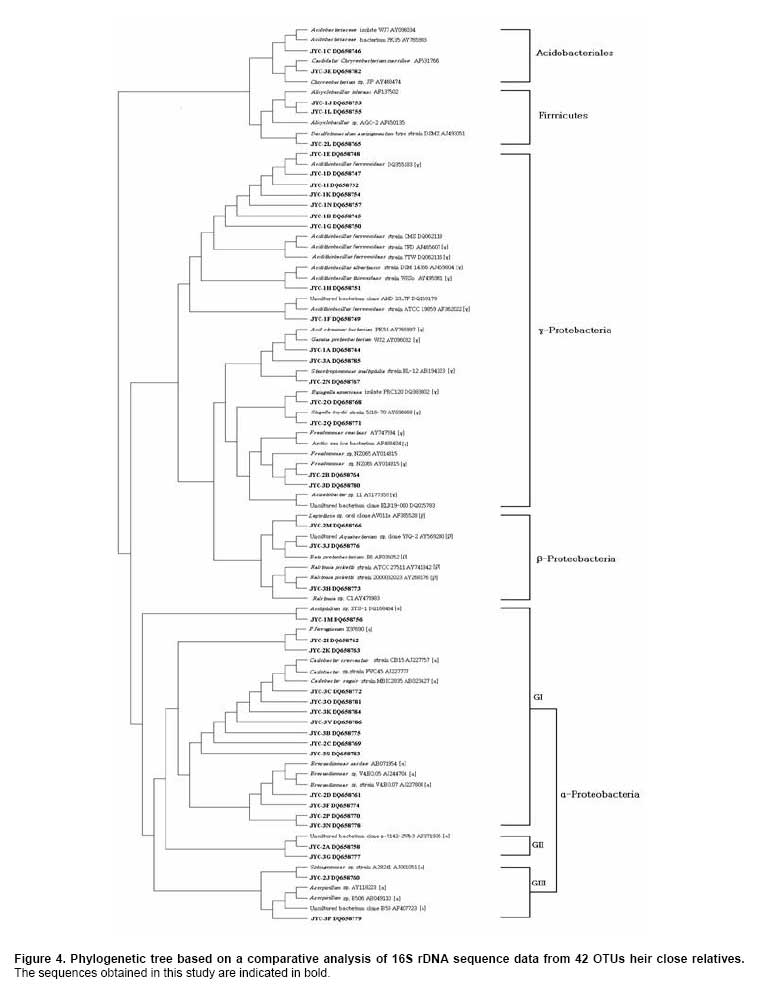
Bioleaching refers to the use of microorganisms for leaching metals such as copper, zinc, uranium, nickel and cobalt from a sulphide mineral. The process involves solubilization of the metals of interest through a series of biological and chemical oxidations of compounds containing sulfur, followed by a metal recovery step (Edwards et al. 1999b; Kelly and Wood, 2000; Verb and Vis, 2001; Baker and Banfield, 2003; Akcil and Koldas, 2006). This widely used technique is an important way to extract metals from low-grade ores (Bosecker, 1997). This technique produces less air pollution and damage to geological formations, but mining companies should be careful of pollution that may generate from ‘acid mine drainage’ (AMD) and contaminate the groundwater (Johnson and Hallberg, 2003). It is known that bacteria play an important role in the formation of AMD. Johnson reported that a wide range of heterotrophic and chemolithotrophic bacteria can be detected in the acid-leaching environments (Dopson et al. 2003). Traditionally, through the culture-dependent methods, Acidithiobacillus ferrooxidans and Leptospirillum ferrooxidans are recognized as the major chemolithotrophic bacteria responsible for acid production in metal-leaching environments (Johnson, 1998; Hallberg and Johnson, 2001). However, many studies have demonstrated that the culture-independent method of analysis reveals a higher degree of diversity than the culture-dependent method when studying the composition of the bacterial community (Edwards et al. 1999a; Bond et al. 2000a; Bond et al. 2000b). Furthermore, many studies on bioleaching have shown that mixed cultures of acidophiles are frequently more efficient in oxidizing sulfide minerals than corresponding pure cultures (Norris et al. 1996; Johnson, 1998). Therefore, it is necessary to understand the microbial community structure in different AMD system, which can improve the metal recovery of bioleaching. To explore the effects of prevailing physico-chemical conditions on associated microbial communities in an abandoned metal mine slag heap, the diversity of bacterial communities from three AMD sites in Baiyin, Gansu Province, China, were investigated using amplified ribosomal DNA restriction analysis (ARDRA). This method involved amplification of the 16S rDNA region, 16S-23S rDNA spacer region or 23S rDNA, followed by digestion with one or more selected restriction enzymes, and has the potential to discriminate bacteria to the species level (Woese, 1987; Amann et al. 1996; Heyndrickx et al. 1996). Sample collection and physico-chemical analyses Three AMD samples (JYC-1, JYC-2, and JYC-3) were collected from three different sites at the Shen-bu copper mine (36º39’00” N; 104º13’35” E), a typical copper mine of Baiyin city in Gansu Province, China, August 2005. These three sites were located near an acid leached slagheap that had been abandoned since 1995. JYC-2 was sampled from an open pool, into which small-amounts of water were continuously being pumped from the underground mine tunnel, and was the largest sampling area of the three sites. JYC-1 and JYC-3 were sampled from two perforated cisterns where only rainwater dropped and flowed. The three sites are isolated and there are no water exchanges between them. The three AMD samples, 50 liters each, were collected and treated within 48 hrs in the laboratory. After being filtered by a vacuum pump through a 0.2 μm nylon filter, the filtrate was used for physico-chemical analyses and the sediments, containing bacteria for DNA analysis, were stored at -20ºC. Physico-chemical analyses of the water samples were performed at the Testing Center of Central South University, Changsha, China. Flame atomic absorption spectrometry (SpectrAA-220FS, Varian, USA) was used to measure metal ions (Okabayashi et al. 2005) and pH was determined using a pH meter (PHS-3C, Leici, Shanghai). Genomic DNA from the samples was prepared (see below) and appropriate 16S rDNA fragments were amplified by polymerase chain reaction (PCR). Fragments of 16S rDNA were then ligated with vectors and a clone library was constructed. The 16S rDNA inserts were reamplified by PCR and digested by Hin6I and MspI (Fermentas, Vilnius, Lithuania). Fragments were separated electrophoretically on a 3% (w/v, solution of agarose in TAE buffer) agarose gel, creating a ‘unique’ banding pattern. The clones corresponding to the unique banding pattern were used for sequencing. The method used for DNA extraction was described previously (Zhou et al. 1996) and was used with minor modifications. The sample material was suspended in 13.5 ml DNA extraction buffer (pH 8.0; 0.1 M phosphate, 0.1 M EDTA, 1.5 M NaCl, 1% CTAB) containing 0.005 mg∙ml-1 proteinase K. The samples were incubated at 37ºC for 1 hr on a horizontal shaker. Thereafter, 1 ml of 20% SDS was added and the mixture was incubated for 1 hr followed by centrifugation at 4000 x g for 5 min at 25ºC. Each supernatant was transferred to a fresh 50 ml centrifuge tube. Following resuspension of the pellets with 4.5 ml extraction buffer by votexing, the suspensions were mixed gently with 0.5 ml of 20% SDS. Samples were then incubated at 65ºC for 15 min, and centrifuged at 4000 x g for 5 min at 25ºC. The resulting supernatants were collected and combined with respective supernatants from the previous separation. The supernatant mixtures were transferred to fresh 50 ml centrifuge tubes, and 1 volume of chloroform was added to each. The mixtures were shaken and centrifuged at 3900 x g for 20 min. The aqueous phases were recovered, and nucleic acids were precipitated for 1 hr by the addition of 0.6 volumes of isopropanol. The DNA samples were pelleted by centrifugation at 10,000 x g for 30 min at 25ºC, washed with 70% ethanol, dried, and resuspended in 200-500 μl autoclaved double distilled water. The quality of genomic DNA was analyzed by agarose gel electrophoresis and a Wizard DNA Clean-Up Kit (Promega, Madison, WI) was used to purify each sample. 16S rDNA amplification and cloning The purified DNA samples were used for amplification of the nearly complete 16S rRNA gene using the two universal primers 63F (5’-GGG CGG WGT GTA CAA GGC-3’) and 1387R (5’- CAG GCC TAA CAC ATG CAA GTC-3’) (Marchesi et al. 1998). PCR was carried out with 0.1 μM of each primer, 200 μM of dNTPs, 2.5 U of Taq DNA polymerase (Perkin Elmer, Norwalk, Conn.) and various amounts of template, in 1 x PCR buffer, adjusted to a final volume of 50 μl with autoclaved double distilled water. The PCR consisted of 30 cycles of denaturation at 95ºC for 1 min (except for the initial denaturation step of 5 min), annealing at 55ºC for 1 min, and extension at 72ºC for 2 min, followed by a final extension at 72ºC for 10 min. The PCR products were visualized by agarose gel electrophoresis and purified by using the Wizard DNA Clean-Up Kit. In order to construct 16S rDNA libraries of the three AMD samples, the purified 16S rDNA fragments were ligated into a pCR2.1-TOPO vector using a TA cloning kit (Invitrogen, Carlsbad, CA). The ligation mixtures were used to transform competent E. coli TOP10 cells (supplied with the cloning kit). Ampicillin-resistant transformants were selected by blue-white screening and grown overnight on plates containing ampicillin (100 mg∙ml-1), IPTG and X-gal. A total of 128 white colonies, from the 3 libraries, were randomly selected. The recombinant plasmids containing 16S rRNA gene fragments were reamplified by PCR using the vector primers M13F (5’- GTAAAACGACGGCCAGTG -3’) and M13R (5’- GGAAACAGCTATGACCATG -3’). PCR amplifications were performed in 50 μl mixtures containing 5 μl of 10 x PCR buffer, 0.5 μM each of both primers, 10 x dNTPs (2 mM each) 5 μl (0.5 U) AmpliTaq polymerase and plasmid DNA. PCR cycling was carried out using a thermal cycler (Mycycler, Bio-rad, USA) as follows: initial denaturation at 94ºC for 4 min followed by 30 cycles of denaturation at 94ºC for 30 sec, annealing at 55ºC for 45 sec, and elongation at 72ºC for 1 min. The amplified rRNA PCR products of the correct size (approximately 1.3 kb) were digested overnight with Hin6I and MspI (Fermentas, Vilnius, Lithuania) at 37ºC. The resulting ARDRA products were separated by gel electrophoresis in 3.0% agarose. The ARDRA patterns were visualized by UV excitation. Jaccard coefficients were computed for all pairwise comparisons of ARDRA banding patterns and dendrograms were constructed using the unweighted pair group mean average method in Molecular Analyst (version 1.1; Bio-Rad, USA). ARDRA banding patterns which were identified were grouped into operational taxonomic units (OTUs), and a representative clone was selected for nucleotide sequence determination, from each OTU. 16S rDNA sequencing and phylogenetic analysis A total of 39 clones, selected from different ARDRA patterns, were sequenced by Sunbiotech Company (Beijing, China). Sequence identification was initially estimated using the BLAST of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/BLAST/). The initial phylogenetic trees were based on all available sequences and were constructed using the DNA distance program Neighbor-Joining with Felsenstein correction in ARB (Smith et al. 1994). After appropriate subsets of 16S rRNA gene sequences were selected, analyzed and aligned with CLUSTAL-X 1.8 (Thompson et al. 1997) based on the initial phylogenetic results, the final phylogenetic tree was generated by MEGA 3.1 (Kumar et al. 2004). Principal component analysis (PCA) was performed using the SYSTAT statistical computing package (version 13.0; SPSS, Inc., Chicago, IL) and consulting protocols described by previous works (Palumbo et al. 2004; Bagwell et al. 2006) for each sampling site. PCA provides a means to separate and group sampling sites based on their complex biogeochemical profiles and community patterns, since it simultaneously considers many correlated variables and then identifies the lowest number to accurately represent the structure of the data (Sharma, 1995). These variables were then linearly combined with the eigenvectors of the correlation matrix to generate a principal component axis. The first principal component axis (PC1) was formed from the original variables with the greatest variance. All subsequent principal components (PC2, PC3, PC4, etc.) were based on the original (high variance) variables that were uncorrelated with the previously defined components. Since each additional principal component had a lower variance than the previous one, most of the variance in the sample data could be accounted for within two or three axes. In this study, PCA provided a means of separating or grouping the AMD samples based on their biogeochemical (pH, temperature, mercury, arsenic, phosphorus, cobalt, magnesium, cuprum, tungsten, zinc, lead, manganese, argentine, sulphur, molybdenum, chromium, iron, aluminum, stannum, calcium, kalium) and biological percentages of OTUs (unique ARDRA patterns) obtained from 16 rDNA parameters. Biogeochemical parameters were entered in Data Editor of SPSS and analyzed by PCA according to the manufacturer's instructions. The analysis was run automatically after entering the data and all data were auto-standardized by SPSS. Rarefaction is a statistical technique for different applications in an ecological context and gives an estimation of the decrease in apparent species richness of a community with decreasing subsample size. In this study, the rarefaction analysis was performed with SigmaPlot software to confirm whether the 16s rDNA library was a reasonable representation of the complete bacterial community or not. An exponential model, y = a x [1-exp (-b x x)], was used with SigmaPlot (version 8.0) nonlinear regression software to fit the clone distribution data. Simpson's diversity index (D) is a simple mathematical measure that characterizes species diversity in a community and it is often used to quantify the biodiversity of a habitat. It takes into account the number of species present, as well as the abundance of each species. Another good measure for moderate discriminating ability is the reciprocal of Simpson’s index (1/D) which is used widely in ecological studies (Magurran, 1988). The value of this index starts with 1 as the lowest possible figure. This figure would represent a community containing only one species. In order to analyze differences in diversity among the sites, we chose to characterize the microbial communities in our AMD samples. The use of 1/D instead of the original formulation of Simpson’s index ensured that an increase in the reciprocal index reflected an increase in diversity (Magurran, 1988; Zhou et al. 2002). Physico-chemical properties of three AMD samples The physico-chemical properties of the three AMD samples are summarized in Table 1. The temperature of the three sites was at 22ºC in the daytime. The pH values ranged between 2.0 and 3.5. All three samples were green, but JYC-3 showed the deepest color. JYC-1 contained the lowest concentrations of all the metals except Fe, and JYC-3 contained the highest concentrations of all the metals except Fe. From each 16S rDNA library, 128 clones were analyzed by ARDRA. The representative ARDRA patterns of the three samples are shown in Figure 1. A total of 39 OTUs were obtained from the different ARDRA patterns. The OTUs were ranked in the order of abundance, and 22, 18 and 14 OTUs were detected in JYC-3, JYC-2 and JYC-1 respectively. Several dominant OTUs were detected in each sample. The calculated rarefaction curve (Figure 2) approached saturation in all of the three samples, indicating that the majority of OTUs in the samples were detected and the diversity of samples in the clone library was almost covered. In sample JYC-1, clones JYC-1B, JYC-1D and JYC-1M were dominant, and accounted for 46.1%, 23.4% and 9.4% of the total, respectively. In sample JYC-2, clones JYC-2C, JYC-2D and JYC-2B represented 41.4%, 12.5% and 12.5% of total OTUs respectively. In sample JYC-3, the OTUs JYC-3A, JYC-3B and JYC-3D (7.8%, 36.7% and 14.0%) were the most abundant. Additionally, JYC-2C was the same as JYC-3B and JYC-2B was the same as JYC-3D (Table 2). There were no mutual ARDRA patterns between JYC-1 and JYC-2, or JYC-1 and JYC-3. The physico-chemical parameters were analyzed by principal-components analysis to assess variability between sites and to identify the parameters driving this variability (Figure 3a). The first two principal components (PC1, PC2) explained 99.37% of the variance, which indicated that little additional information could be obtained by further principal components (PC3 or more). Therefore, from the PCA results, the first axis suggested that three sites were distant from each other in terms of their physico-chemical properties. The PCA results for biological parameters suggested that the first two principal components explained 95.5% of the variance. According to the PCA (Figure 3b), the overall bacterial community structures in JYC-2 and JYC-3 were close together and significantly different from those observed in JYC-1. To quantitatively measure diversity in the samples, we used the inverse of Simpson’s index (1/D), which is sensitive to the level of dominance in a community. The reciprocal of Simpson’s index (1/D) effectively distinguished the three samples from each other and these calculations of diversity shown that the diversity of JYC-1 sample (1/D:2.88) was lowest and the diversity of JYC-2 sample (1/D:4.17) was lower than the diversity of JYC-3 sample (1/D:7.04). Sequence data and phylogenetic analysis of 16S rDNA sequences Thirty-nine clones, from all three samples, were re-amplified and sequenced, including clones that had the same ARDRA patterns from different samples. All successful sequences were submitted to Genbank, and the accession numbers listed in Figure 4. The phylogenetic tree was constructed by the bootstrap neighbor-joining method with the sequences from all known and candidate divisions (Figure 4). Most of the 16S rDNA clones were closely related to the candidate divisions that belonged to α, β and γ-Protebacteria. There were also two other divisions: one clone for Acidobacteriales and three clones for Firmicutes respectively. The most abundant clones in the samples fell into the γ and α subclass of the Protebacteria, which were closely related to A. ferrooxidans and C. crescentus. Only 14 OTUs were detected in sample JYC-1. Sequence analysis revealed that the majority of the clones in JYC-1 were affiliated with the γ-Protebacteria (85.1% of the total clones). Two dominant clones, JYC-1B (46.1%) and JYC-1D (23.4%), were both closely related to A. ferrooxidans (99% and 100% similarity). Clone JYC-1M (9.4%) was affiliated with α-Protebacteria, which was associated with Acidiphilium sp. XTS-1 (DQ168464.1, unpublished). Clone JYC-1A (4.7%) and clone JYC-1H (3.9%) were also affiliated with γ-Protebacteria and showed similarity of 98% with Gamma proteobacterium WJ2 (Hallberg and Johnson, 2003) and 100% with Acidithiobacillus albertensis (Bryant et al. 1983). Clones JYC-1C (3.1%) and JYC-1J (2.4%), which did not fall into Protebacteria were closely related to Acidobacteriales and Firmicutes. Data analysis of JYC-2 and JYC-3 The phylogenetic distributions of JYC-2 and JYC-3 were similar and they shared five OTUs with each other. Clones JYC-2C (41.4%) and JYC-3B (36.7%) were the most abundant sequences recovered from each site, and were 99% similar to C. crescentus (Nierman et al. 2001). In the case of other α-Proteobacteria, Pseudomonas reactans (AY747594.1 unpublished) was less dominant than C. crescentus in JYC-2 (12.5%) and JYC-3 (14.0%), which was named JYC-2B and JYC-3D. JYC-2M (3.1%) in JYC-2, JYC-3J (5.5%) and JYC-3H (2.4%) in JYC-3 were closely related to Leptothrix sp. (Paster et al. 2002), Aquabacterium sp. and Ralstonia pickettii, respectively. At the genus level, the similarities and differences among the three sites were pronounced. Clones that were similar to A. ferrooxdans were most dominant in the samples of JYC-1 representing 76.5% of the total clones, but they did not occur in JYC-2 and JYC-3. Clones similar to C. crescentus were clearly the most dominant in JYC-2 and JYC-3 representing 41.4% and 36.7%, respectively. Most of the clones from JYC-1 clustered with γ-Protebacteria, while many clones from JYC-2 and JYC-3 clustered with α-Proteobacteria. There were few clones in JYC-1 (JYC-1C, 3.1%) and JYC-3 (JYC-3E, 5.5%) which fell into Acidobacteriales, but no such clone was detected in JYC-2. JYC-1J (2.4%), JYC-1L (1.6%) and JYC-2L (3.9%) appeared to be affiliated with Firmicutes. β-Proteobacteria was detected in JYC-2 (JYC-2M, 3.1%) and JYC-3 (JYC-3J, 5.4% and JYC-3H, 2.4%), but not in JYC-1. The aim of this study was to use ARDRA analysis to characterize the diversity and community structure of AMD and to assess the differences between samples that varied in their pH and metal contents, factors which were anticipated to influence the phylogenetic diversity of microbial communities. The sequences prepared in this study represented a wide range of bacterial phylogenetic groups. In general, bacteria belonging to Acidobacteriales, Firmicutes and Protebacteria were found. A comparison of these results with previously reported data (Baker and Banfield, 2003) highlighted the absence of Nitrospira from these samples. Phylogenetic analysis of the sequences indicated that there were differences in diversity and the relative incidence of particular phylogenetic groups among the three samples. Sequences of clones recovered from JYC-1 belonging to the γ-Proteobacteria were the most abundant (almost 85.1%). These bacteria are typically found in low pH environments (Bond et al. 2000a; Bond et al. 2000b; Burton and Norris, 2000; Dopson et al. 2003; Gonzalez-Toril et al. 2003; Okabayashi et al. 2005). In contrast, the most abundant clones (60.2 and 52.4%) in samples JYC-2 and JYC-3 belonged to α-Proteobacteria. These organisms have previously been found in heavy metal-contaminated soils (Sandaa et al. 1999). To summarize, pH and soluble metal concentrations exert a considerable influence on the phylogenetic diversity of microbial communities in AMD. We used the inverse of Simpson’s index (1/D), which is sensitive to the level of dominance in a community (Magurran, 1988) to measure diversity in the samples. The reciprocal of Simpson’s index (1/D: 2.88 to 7.04) effectively demonstrated diversity of bacterial communities in AMD studied here were lower than other ordinary environments, such as surface soils (Zhou et al. 2002), and marine sediments (Ravenschlag et al. 1999). Even at 5 to 12 cm below the sea floor at a depth of 3,814, the 1/D value of Actinobacterial diversity was 51 (Stach et al. 2003). It is noteworthy that the AMD environments we characterize here had severely low microbial diversity indices. Metals at high concentration and low pH are toxic to microorganisms (Bruins et al. 2000). The reason for the low microbial diversity indices appeared in our study could be the high ion concentrations and low pH. Furthermore, diversity of communities associated with the sites described here were distinct. For example, 1/D values were 2.88 and 7.04 respectively in JYC-1 and JYC-3 samples. The microbial communities among these sites were complicated assemblages that were influenced by a number of variables. The PCA data showed that the diversity of JYC-2 and JYC-3 were similar and they were both distinct from JYC-1. The three sites were distinct from each other with respect to their physico-chemical properties. These results indicate that there could be some key physico-chemical properties that influenced the structure of the bacterial communities. For sample JYC-1, the pH value (pH 2.0) and metal concentrations, except iron, were the lowest while the iron concentration was the highest, of the three samples. According to previous reports (Bryant et al. 1983; Schrenk et al. 1998; Baker and Banfield, 2003), the low pH value and iron concentration were important parameters for the presence of many acidophilic microorganisms. Iron is the most biologically important metal for the growth of A.ferrooxidans. Many sequences in the library of JYC-1 were related to sequences previously found in AMD (Baker and Banfield, 2003). For example, the most abundant microbial components in this sample were identified as A.ferrooxidans,Acidiphilium sp., and A. albertensis. But in JYC-1, the low bacterial diversity according to ARDRA analysis was consistent with the results of Baker and Banfield (2003) in a similar environment. Sequences attributable to Leptospirillum spp. were not encountered in our clone libraries, even though Leptospirillum spp. have been observed to be more abundant than A. ferrooxidans in another AMD system (Bond et al. 2000a). This implicates the toxicity of soluble metals in the samples studied here. Though JYC-1 had the lowest metal concentration of the three samples, the concentration was higher than those reported in previous AMD studies (Dopson et al. 2003; Gonzalez-Toril et al. 2003; Okabayashi et al. 2005). Johnson reported that Leptospirillum showed growth in concentrations of Cu lower than 5 mM, which was much lower than A. ferrooxidans (tolerant to 800 mM) (Johnson et al. 1992; Dew et al. 1999). No normal acidophilic bacteria were detected in the JYC-2 and JYC-3 samples. Some bacteria were first detected in AMD, such as sequences affiliated with C. crescentus, Pseudomonas sp., Leptothrix sp., Aquabacterium sp. and Ralstonia pickettii. Such variations could be consistent with the occurrence of higher soluble metal concentrations and higher pH. Our data indicated that JYC-2 and JYC-3 had much higher soluble metal concentrations than JYC-1, especially for Zn, Cu, Mg and Ca. In addition, a higher pH value (pH 3.5) was measured in the two samples. The bacterium C. crescentus is known for the distinctive ability to live in low-nutrient environments, a characteristic of most heavily metal-contaminated sites. Other clones affiliated with Pseudomonas sp., Leptothrix sp., and Ralstonia pickettii had all been reported to be tolerant of high concentrations of soluble metals (Dressler et al. 1991; Johnson et al. 1992; Goris et al. 2001). For example, Ralstonia exhibited resistance to at least 1200 mg/L of Cu (Konstantinidis et al. 2003). The absence of normal acidophilic bacteria in JYC-2 and JYC-3 may have been caused by the high concentration of soluble metals. The bacteria mentioned above were reported as neutrophiles, but the pH values at our sampling sites were considerably lower than those previously reported in the literature (Dressler et al. 1991; Johnson et al. 1992; Goris et al. 2001). The reason for this discrepancy could be the intense evaporation of water in sampling days due to drought, which led to a lower pH value of the pool water than on normal days. Since the bacterial strains have not been isolated, their physiological role in the ecology will remain uncertain, so further investigation should be concentrated on isolating these bacteria. Thanks are expressed to Weiliang Chao, Department of Microbiology, Soochow University, Taipei, for invaluable suggestions. We are grateful to Shuyu Wang, School of Public Health, Sun Yat-sen University, Guangzhou, China, for critically revision of this manuscript.
Note: Electronic Journal of Biotechnology is not responsible if on-line references cited on manuscripts are not available any more after the date of publication. © 2008 by Pontificia Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej08001f1.jpg] [ej08001f3.jpg] [ej08001f2.jpg] [ej08001t1.jpg] [ej08001t2.jpg] [ej08001f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}