 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Electronic Journal of Biotechnology, Vol. 13, No. 2, March 15, 2010 TECHNICAL NOTE Development and application of high-throughput amplified fragment length polymorphism technique in Calluna vulgaris (Ericaceae) Thomas Borchert*1 , Inka Gawenda2 1 Leibniz-Institute of Vegetable and Ornamental Crops,
Department of Plant Propagation,
Kuehnhaeuser Str. 101,
Erfurt, Germany
E-mail: borchert@erfurt.igzev.de Financial support: The results were obtained within a third-party funded enterprise (German Federal Ministry of Education and Research, grant PGI-06.01-28-1-43.038-07). The experiments were conducted at the IGZ. Received July 14, 2009 / Accepted October 23, 2009 Code Number: ej10021 Abstract Calluna vulgaris is an important ornamental crop of the horticultural industry in Europe. In order to improve breeding of this species, especially of the most important trait of ‘bud-flowering’, the implementation of molecular techniques that allow rapid, reproducible and efficient screening of whole segregating populations e.g. for molecular marker and mapping approaches is a requirement. We therefore aimed to introduce the powerful tool of amplified fragment length polymorphisms (AFLP®), a widely and successfully applied method, into our methodological assortment. As an essential prerequisite, the isolated DNA should be of adequate quality which is a common obstacle when dealing with woody species and their interfering secondary components/metabolites. The results of screening different and modified DNA isolation protocols are described. As the outcome of our evaluations of reaction conditions during the AFLP® procedure, we circumstantiate a functional protocol ranging from DNA extraction to visualization of AFLP® banding patterns for the woody crop C. vulgaris. This method is suitable for high throughput genetic applications and may even be transferable to other species. In addition, costs are reduced by reasonable reagents and multiplexing assays. Keywords: DNA content, mapping, marker-assisted selection, ornamental, plant tissue.
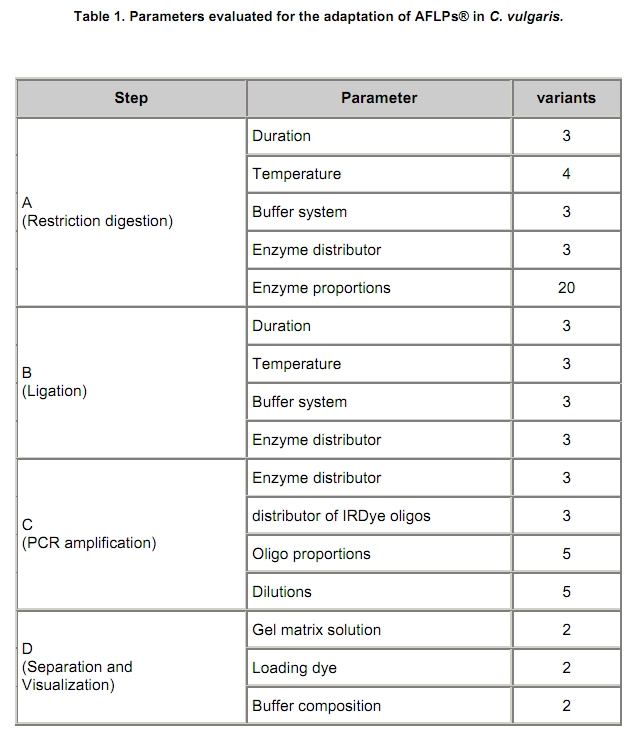
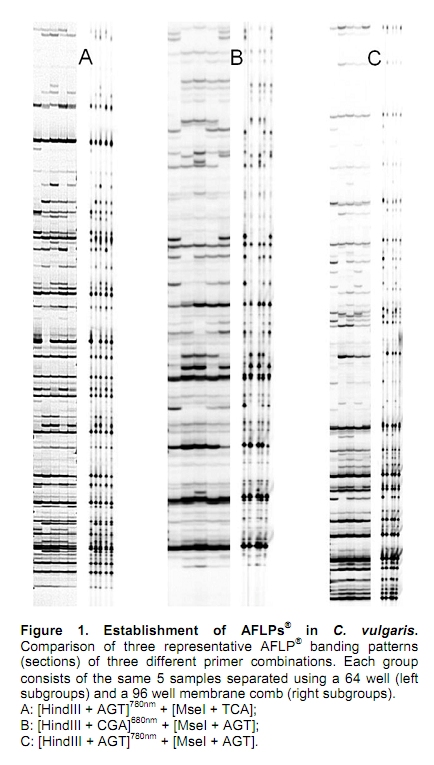
Recently, we reported a DNA isolation method for the ornamental crop C. vulgaris suitable for randomly amplified polymorphic DNA (RAPD) and inter simple sequence repeat (ISSR) analyses (Borchert et al. 2008). However, this method incorporates potentially harmful phenol/chloroform extraction steps and requires approximately 14 hrs of working time for 24 samples (from tissue grinding to quantification and qualification). Moreover, RAPD and ISSR techniques are not adequate e.g. regarding reproducibility especially for mapping of traits. Therefore, we aimed at improving our method by simplifying the DNA isolation protocol for C. vulgaris and establishing amplified fragment length polymorphisms (AFLP®) methodology. Both methods should comply with high-throughput (htp) requirements in terms of time- and cost-efficiency. AFLPs® require high-quality DNA. The isolation of such DNA of herbaceous and woody plants is commonly hindered due to the accumulation of polyphenolic and polysaccharide compounds that interfere during subsequent processing of the DNA. Several manual protocols based on different assays to eliminate these contaminants (e.g. CTAB, silica; (Rogstad, 2003)) are available for such cases. However, for each species the adaptation of the isolation method is usually required. Since its initial description by Vos et al. (1995), AFLPs® were successfully adapted to different species of e.g. the Erica family (Rhododendron: de Riek et al. 1999; Vaccinium: Albert et al. 2003); Rubus: Graham et al. 2004; Gaylussacia: Pooler et al. 2006) and Osteospermum (Asteraceae: Gawenda and Debener, 2009). As a result of the technological progress, this marker system is currently and routinely applied for genetic, evolutionary and ecological studies (Meudt and Clarke, 2007). The AFLP® technique consists of at least four steps: (A) restriction digestion of genomic DNA, (B) ligation of adapter oligos, (C) selective amplification of generated fragments by restriction site specific PCR and (D) separation and visualization of amplified fragments. Each step typically requires adaptations regarding concentrations and/or dilutions of template, enzymes and reagents. Step A and B may be combined in one reaction, step C is separated into two discrete PCR runs. Distinct adjustments are required for the primer combinations used for research purposes, since the specificity of the oligo nucleotides is defined by additional (= selective) nucleotides at the 3’-end. The stringency of fragment selection directly influences the amount of fragments visualized in step D and the subsequent comfort and efficiency of automated analysis. Furthermore, the DNA content of the species under investigation is known to influence the amount and quality of AFLP® banding patterns (Fay et al. 2005). We recently determined the DNA content of C. vulgaris to be 1.18 pg / 2C (Borchert et al. 2009), which is a relatively low value. For such a case, the number of analyzable bands may be increased by decreasing the number of selective nucleotides. According to Fay et al. (2005), who reported a primer combination of (+3/+3) selective nucleotides to be functional in species with a genome size between 0.6 and 24.0 pg / 2C, equivalent primer combinations may be expected to be functional in C. vulgaris. Prior to AFLP® establishment, several methods for isolation of genomic DNA were comparatively screened. The AFLP® technique was subsequently adjusted with regard to the above mentioned criteria and the laboratorial circumstances (LiCor DNA4300S analysis system, capable of detecting fluorescence-labeled DNA during polyacrylamide gel electrophoresis). Materials and Methods The following isolation methods were evaluated with fresh shoot foliage of the same C. vulgaris back-crossing progeny individuals (n = 2 x 2): #1: manual method (Borchert et al. 2008), #2: Plant Genomic DNA Mini Kit (Avegene), #3: manual method (Chen and Ronald, 1999), #4: Power Plant Isolation Kit (MoBio), #5: DNeasy Plant Mini Kit (Qiagen), #6: NucleoSpin Plant II (Macherey-Nagel). The protocols were conducted (i) using the original buffers and recommendations and (ii) using modified buffers which were supplemented with 0.5% (v/v) beta-mercaptoethanol, 1.25% (w/v) sodiumsarcosyle and 2% (w/v) Polyvinylpyrrolidone-40. These modifications were deduced from our experiences regarding DNA extraction, which is commonly complicated by secondary metabolites i.e. phenols and carbohydrates. The buffers of the Macherey-Nagel Kit were not modified, since it already included two different buffer systems (SDS vs. CTAB). The reagents for AFLP® analysis were obtained from New England Biolabs (HindIII, MseI, T4 Ligase and appropriate buffers), metabion (Taq Polymerase, oligos, infrared dye labeled oligos (IRDye®) and LI-COR (KB + 6.5% matrix solution). PCR conditions and levels for testing necessary modifications in C. vulgaris were primarily adopted from Vos et al. (1995) and some modifications mentioned by Gawenda and Debener (2009). The parameters evaluated for modifications are shown in Table 1. All reactions were conducted using the original manufacturer’s protocol of the respective step regarding buffer concentrations and temperature ranges. Typically, only standard modifications of the sub steps were tested e.g. ‘1 hr’ vs. ‘4 hrs’ vs. ‘over night’ within step A (duration of restriction digestion). Results and Discussion Genomic DNA was isolated by different methods, separated by agarose gel electrophoresis and quantified vs. Lambda DNA (Fermentas) and spectrometric methods. In comparison to method #1, our established protocol, only the Qiagen DNeasy Plant Mini Kit (#5) resulted in genomic DNA of highest quantity and a quality lacking any contamination. Protocol #2 was not capable to completely eliminate RNA, protocol #3 did not sufficiently remove carbohydrates. Protocols #2, #3, #4 and #6 led to DNA shearing in comparison to protocols #1 and #5. Surprisingly, the chemical modifications described did not result in significant improvements of DNA quality and quantity in any of the methods tested in comparison to the standard manufacturer’s protocols although this was expectable from our previous experiences with this woody ornamental regarding both DNA and RNA isolation. Furthermore, other authors (Dhanaraj et al. 2004) already described the requirement of these supplemental chemicals for the extraction of floral bud RNA of Vaccinium, a species of the same plant family (Ericaceae). The overall yield of the Qiagen kit (ranging from 10 to 50 ng/µL) was clearly and reproducibly lower than the concentration achievable by method #1 (ranging from 30 to 120 ng/µL). Furthermore, both types of DNA proved its repeatable suitability for AFLP® analysis e.g. regarding banding pattern reproducibility from independent DNA isolations of the same genotype. However, taking into account costs and time requirements for the isolation procedure, the Qiagen DNeasy Plant Mini Kit was chosen as an adequate isolation method for C. vulgaris. As a result of the experiments outlined by Table 1, the following modified protocol shaped up to be successful to generate reproducible AFLP® banding patterns. This protocol was capable of the simultaneous visualization of fragments generated in multiplexed PCR reactions. Step A: Restriction digestion. In a total volume of 25 µL containing 1 X Buffer 2 (NEB), 1 X BSA (NEB), 150 ng genomic DNA were double digested by HindIII (5 Units, NEB) and MseI (3 Units, NEB) over night (at least 12 hrs) at 37ºC. Step B: Ligation. 5.0 µL of a ligation mix (containing 0.5 µM HindIII adapters, 5 µM MseI adapters, 1 Unit T4 DNA Ligase (NEB), 1 X T4 Ligase reaction buffer (including ATP, NEB)) were added to the 25.0 µL restricted DNA of step A and incubated for 4 hrs at 37ºC. Step C: Selective amplification. PCR amplification was separated into two steps. In step one (C1), all fragments containing both the HindIII and the MseI restriction site on opposite ends were amplified by non-selective and non-labeled primers (+0/+0) complementary to adapter sequences. In a second step (C2), selective primers were applied that furthermore significantly reduce the amount of fragments. Here, the HindIII primers were labeled with IRDyes® (680 nm or 780 nm, respectively) for subsequent visualization by laser excitation. Step C1: In a total volume of 50.0 µL the following reagents were used for amplification: 5.0 µL of DNA of Step B, 75 ng of [HindIII + 0] and [Mse + 0] primers each, 10.0 mM dNTPs, 1 X buffer (metabion), 5 Units Taq Polymerase (metabion). The PCR conditions differed from Vos et al. (1995) in the annealing time (30 sec instead of 1 min) and temperature (60ºC instead of 56ºC). 30.0 µL of amplification products were diluted 1:20 in 1 X TE buffer and can be stored by -20ºC. Residual PCR products were checked on a 1% agarose gel for resulting smear due to different fragment sizes. Step C2: In a total volume of 10.0 µL the following reagents were used for selective amplification: 2.5 µL 1:20 diluted DNA of Step C1, 15 ng [MseI + Nx] primer, 0.2 mM dNTPs, 1 X buffer (metabion), 0.25 Units Taq Polymerase (metabion). In a mono-reaction, only one IRDye®-labeled primer was used ([HindIII + Nx], 5 ng); in a dual reaction, both the 680 nm and the 780 nm laser scanning system and appropriately labeled IRDye® primers were used in parallel (4 ng each). Step D: Separation and visualization. 25 cm AFLP® gels of 0.25 mm thickness (LI-COR KB+ 6.5% gel matrix, 150 µL 10% APS, 15 µL TEMED) were hardened for at least 4 hrs and used to separate PCR fragments. Samples resulting of Step C2 were diluted 1:11 with loading dye (98% formamide, 10 mM EDTA, 0.05% pararosaniline) and denaturated 3 min at 94ºC. 0.5 µL were loaded onto a sample-loading tray. A 96 well membrane comb (LI-COR part# 9842-168) was dipped into the samples for no longer than 5 sec to absorb the liquid. After the Pre-Run (750 V, 20 mA, 15 W, 45ºC, 20 min), the membrane comb is inserted between the glass plates according to the manufacturer’s protocol. After 1.5 min of the Collection-Run (1500 V, 35 mA, 35 W, 45ºC, 3 hrs 30 min) the comb is removed. Alternatively, 0.5 µL of each sample were loaded in between the tooth of a 64 well plastic shark tooth comb. As already described by Myburg et al. (2001), AFLP® gels can be loaded at least twice. Fragment separation quality is reduced only marginally between the two runs on the same gel matrix (not shown). Figure 1 comparatively shows representative AFLP® banding patterns of five C. vulgaris genotypes in the range of up to 300 bp generated by separation using the 64 well comb (left lanes) or the 96 well membrane comb (right lanes) as described above. The discriminative power of the 96 well htp assay is not reduced in comparison to the 64 well assay, since all polymorphisms are readily detectable. Typically, bands are reduced to dots, but even the fragment intensities are reproducible between both gel loading variants. Thus, the use of a 96-sample membrane comb is applicable and in combination with automated analysis software hundreds of AFLP®-markers can be obtained in a short time. The number of AFLP® fragments was expected to be low, due to the small genome size of C. vulgaris (1.18 pg / 2C, Borchert et al. 2009). The number of fragments may be increased by application of primers with two or even only one selective nucleotide in step C2. However, PCR amplification of the selective primer combination [HindIII + N2] and [MseI + N2] resulted in a tremendous increase of fragments that outweighed data interpretation. Thus, we assume resolution and discriminative power of AFLPs® in C. vulgaris to be adequately effective by application of three selective bases at the 3’-end of each the HindIII and MseI primer (+3/+3) or (+3/+2) at most. Using both excitation systems in parallel, a 96 well paper comb and conducting two runs with one gel, 384 different reactions can be screened within 7 hrs of apparatus running time and approximately 1 hr of working time (gel preparation and sample loading) using the protocol described above. Considering the costs for the reagents our protocol results in expenses of less than 12 EUR per sample. Of these, ~21% alone are made up of the expenditures for DNA isolation. A switch towards a molecular marker system capable of such high throughput and reproducibility in comparison to e.g. RAPDs (Bagley et al. 2001) was necessary for our prospect genetic applications. These aim to improve the breeding of the economically important ornamental crop C. vulgaris. Thus, the low-cost htp protocol described above can be applied for screening of molecular markers for relevant traits and mapping approaches since it is capable of screening many genotypes simultaneously in a short time span. Acknowledgments The authors thank Annette Hohe (IGZ) for managing the project funding and for participating in drafting this manuscript and Marcus Linde (LUH) for his academic support regarding methodology establishment. References
Note: Electronic Journal of Biotechnology is not responsible if on-line references cited on manuscripts are not available any more after the date of publication Supported by UNESCO / MIRCEN network Copyright © 2010 by Pontificia Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej10021f1.jpg] [ej10021t1.jpg] |
| |||||||||

{kind=link}
{kind=link}