 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
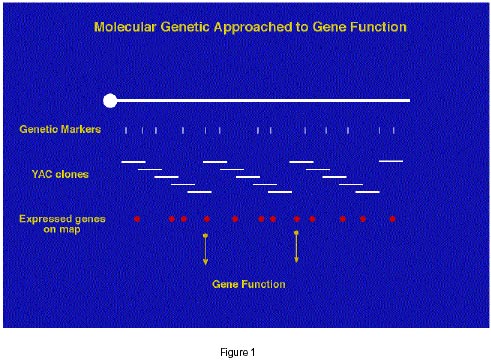
Electronic Journal of Biotechnology, Vol. 1, No. 1, April, 1998 INVITED REVIEW ARTICLE Global analysis of gene function in mammals: Integration of physical, mutational and expression strategies John Schimenti The Jackson Laboratory, Bar Harbor, ME 04609, USA Tel: 207-288-6402 E-mail:jcs@jax.org Code Number: ej98006 The field of Genetics is undergoing tumultuous change after nearly a century of standard approaches to genetic analysis. The Human Genome Project is providing tools and technologies that are changing the ways that we pursue an understanding of gene function, which is the underlying goal in modern and traditional genetics. In this paper, I overview the directions of the genome project as they relate to gene function analysis in mice and humans, and how various modern technologies are coalescing to address this in a powerful way. Keywords :Gene expression, ESTs, Mutagenesis, Genome, Functional genomics The long term goal of the genome project is to define and understand all the genes specifying the making of a human being. Ultimately, this knowledge will improve the human condition through genetic diagnostics, gene therapy, and improved understanding of biochemical and physiological processes. The benefits of the genome project are already being realized. High resolution genetic maps have been generated for humans and other experimental organisms, leading to several notable successes in positional cloning of disease genes. Over the past few years, the completion of genetic maps has been transitioning to physical mapping of genomes and to the characterization of their functional content. The sequencing of Expressed Sequence Tags (ESTs) is relentlessly moving towards the identification of nearly all transcribed genes in mouse and humans (Adams et al., 1995; Hillier et al., 1996) . These ESTs are being placed onto the genetic and physical maps, further facilitating the process of positional cloning. New technologies are being developed to enable the simultaneous analysis of the expression patterns of thousands of genes on a large scale and under different circumstances (Chee et al., 1996; DeRisi et al., 1997; Schena et al., 1995) . The physical mapping stage will culminate in the complete sequencing of the human genome, and perhaps the genomes of other complex model organisms such as mouse, Drosophila and C. elegans. The genomes of several bacteria as well as baker's yeast have been completed. The most difficult challenge now lies in devising ways to utilize the vast amount of information gathered in the Genome Project to understand how organisms develop and function. Even after all the genes are identified, and their transcription patterns revealed, and the entire genomic sequence known, the function of the genes will still remain unclear (Miklos and Rubin, 1996; Oliver, 1996) . The problem of elucidating function for all the 100,000 or so genes in humans/mammals has spawned a new area of research that is being called "functional genomics." The goal of the functional genomics effort is to determine the biological function of a genome using strategies that will ultimately coalesce with the genetic maps, physical maps, DNA sequence, and gene transcription patterns. Molecular characterization of genesPart of the functional genomics field deals strictly with nucleic acids. The philosophy behind it is that much can be learned about the function of genes by simply isolating them, evaluating their transcriptional pattern during development, and predicting molecular function by inference (based on known functions of related genes or protein motifs). The cornerstone of molecular biological approaches in the genome project is the identification of expressed genes by brute force sequencing - the generation of Expressed Sequence Tags (ESTs). Several companies, independent research groups, and large efforts such as the IMAGE consortium (http://www-bio.llnl.gov/bbrp/image/image.html) have been performing single pass sequencing of hundreds of thousands of cDNA clones from many different sources. Collectively, the data represent most of the transcribed genes in humans and model organisms such as mice. The next step in this area of work will be to collect full length sequence from a non-redundant subset of the clones. The EST projects are key in the next step of molecular biological analysis of gene function - to ascertain the patterns of expression of all the genes. This has required the development of sophisticated new technologies. These technologies are required for the following reason. Unlike the old ways of measuring gene expression, such as Northern blotting, RT-PCR or in situ hybridization, which are designed to assay expression of one gene at a time, the new technologies are designed to analyze expression of thousands of genes simultaneously. One method is called SAGE (serial analysis of gene expression), which involves sequencing templates consisting of short oligomers of many genes, whereby the number of times a given gene "tag" is sequenced is reflective of the degree to which the corresponding gene is expressed (Velculescu et al., 1995) . More powerful, however are the "gene chips" being developed (Chee et al., 1996; DeRisi et al., 1997; Schena et al., 1995) . In this technology, DNA corresponding to thousands of genes are arrayed on small matrices ("chips"), and probed with labelled cDNA from a tissue of choice. The information from an experiment is then read by a device to download into a computer. Both technologies can also be exploited to determine what genes are up- or down-regulated under different conditions or disease states. As I will discuss later, the characterization of expression patterns is critical for linking phenotypes with candidate disease genes. Physical approachesThe ultimate goal of the physical mapping aspect of the genome project is to determine the entire sequence of the human genome and other model organisms. Because the genome is so large, this must be done in an organized fashion by compartmentalizing. Ideally, this would involve sequencing of ordered clones spanning each chromosome. The first step was to generate linkage maps of the mouse and human genomes, and the same is being done for rat. Basically, the strategy has been to identify several thousand polymorphic microsatellite markers that could be typed by PCR. Comprehensive linkage maps for mice and humans were generated within the last few years (Dib et al., 1996; Dietrich et al., 1996) , leading to a revolution in mammalian genetics. Using the genetic map as a scaffold, Yeast Artificial Chromosome (YAC) contigs spanning much of the human and mouse genomes were (and are still being) constructed (Cohen et al., 1993) ; (http://www-genome.wi.mit.edu/cgi-bin/mouse/index). However, YACs are problematic in that they are prone to undergo rearrangement inside yeast, and are often chimeric in the sense that individual YACs will contain sequences in the genome derived from multiple chromosomes. Finally, they are difficult to isolate away from yeast DNA, and are so large as to be unmanageable for many tasks. Cosmids are much smaller (about 40 kb), but too small to be used for constructing contigs across the genome in an efficient way. A happy medium appears to be Bacterial Artificial Chromosomes (BACs), which are of a more manageable size (in the 100-200 kb range) and appear to be much more stable and not as subject to chimerism. At present, BAC contigs across the mouse or human genome do not exist, but it is likely that efforts will be funded to perform this task. What is especially attractive about the route of generating a BAC contig across the genome is that it will be useful in the interim while sequencing is being performed. As discussed below, an ordered contig of BACs would greatly reduce the effort required in positional cloning. GeneticsIt is ironic that what we are now calling "functional genomics" is really just a new word for "genetics." The goal of both disciplines is to understand what genes do. The only difference is that functional genomics seeks to do this on a larger and faster scale. The basic tool of a geneticist is allelism - a difference in genetic composition that manifests itself in a visible, measurable way. Classically, this would involve a mutant. Indeed, this still applies today. In humans and mice, naturally occurring mutations often take the form of diseases. There are a variety of spontaneous mutants in mice that confer all imaginable types of phenotypes, from coat color alterations to neurological effects. The molecular basis for many mutations has been determined in many instances through positional cloning. This still involves constructing high generation genetic maps through linkage analysis : recombinational mapping. Unfortunately, these exploits can occupy the entire attention of a laboratory for several years, a period in which little biological information is obtained. The ability to manipulate mammalian genes in vitro in embryonic stem cells, combined with the generation of transgenic mice harboring DNA of these ES cell in their germ cells, has advanced the mutagenesis of specific genes (targeted mutagenesis). Similarly, the application of gene traps to ES cells (which is essentially insertional mutagenesis akin to transposon tagging in flies, bacteria and plants) has endowed the mouse with a powerful gene-based means of mutagenesis: that is, it has become relatively simple to generate a mutation of any known molecularly-identified gene. However, the mouse field has seriously lagged in one major respect: the ability to perform systematic, phenotype-based screens on a scale large enough to reach the level of saturation. The nature of flies enables the application of chemical, deletional and insertional mutagenesis approaches to identify mutations in genes that cause specific phenotypes in the whole organism. Mutagenesis screens have been performed on a scale that is sufficiently large to ensure recovery of an array of mutations that elicit a particular phenotype. Collections of such phenotype-selected mutants in flies have had a major impact in the elucidation of gene function in developmental pathways. Importantly, the hierarchy of gene (inter)actions or ordering of genes in functional pathways could be established without prior knowledge of the molecular nature of the gene product. The Nobel Prize in Medicine was awarded to Weishaus, Rubin and Nusslein-Volhard for pioneering this type of work in Drosophila melanogaster. The experimental obstacles posed by mammalian model systems have made it impractical to efficiently pursue mutagenesis strategies as in invertebrates. While the mouse will never be as amenable to comprehensive phenotype screens as flies, recent progress has nevertheless provided the tools to perform in vivo mutagenesis that is efficient enough to enable phenotype screens. One type of mutagen that has been exploited is radiation. Ionizing radiation has been used to generate mutations in mice for quite some time, ultimately to create "deletion complexes" around several visible loci (Holdener-Kenny et al., 1992; Lyon and Morris, 1966; O'Brien et al., 1996; Rinchik, 1994; Russell et al., 1995; Russell et al., 1982) . However, this process is very inefficient and labor intensive, and requires visible markers to select the deletions. Two procedures have recently been developed to create large chromosomal deletions in ES cells. One method uses Cre-LoxP site specific recombination to remove sequences between two targeted loxP sites, thereby resulting in a single, precise deletion (Ramirez-Solis et al., 1995) . The other method is designed to produce deletion complexes by irradiation of ES cells containing a targeted thymidine kinase gene (You et al., 1997; You et al., 1997) . This makes it possible to derive mice bearing sets of nested deletions anywhere in the genome. A drawback of deletions is that they remove multiple genes, complicating the analysis of a phenotype. To generate single locus mutations, other mutagens, namely those that induce small intergenic lesions, are employed. In flies, the chemical EMS (ethyl methane sulfonate) is used routinely to create single gene mutations or new alleles of previously mutated genes, while in the mouse and zebrafish, the most potent and commonly used chemical mutagen is N-ethyl-N-nitrosourea (ENU) (Justice and Bode, 1986; Shedlovsky et al., 1988; Vitaterna et al., 1994) . With the technology in place to perform high efficiency point mutagenesis in mice and to make large deletions, it is now possible to conduct "region specific mutagenesis in a comprehensive way. The general strategy of region-specific saturation mutagenesis presented in this review has been extensively utilized in Drosophila (Ashburner, 1989) . This approach provides a tool for identifying and characterizing genes in a particular chromosomal subregion in a simple, two generation breeding scheme. The basic principle is to cross mice that have been mutagenized by ENU to mice that bear a known chromosomal deletion. The offspring that inherit the deletion from one parent and an altered/mutated gene from the other parent will display a mutant phenotype. This simple breeding scheme allows phenotypic characterization of a large number of mutagenized gametes - sufficient to reach theoretical "saturation," such that a mutation in every gene in the region should have been produced and scored for a novel phenotype. An attractive aspect of a region-specific mutagenesis is that a panel of nested deficiencies, when available, can be used for fine mapping of a newly induced mutation by complementation analysis. Such classical mutagenesis strategies, in part, were responsible for making Drosophila melanogaster a premier model for developmental and molecular genetic analyses of higher organisms. From a geneticists point of view, mutations are absolutely required to truly understand the function of a gene in the context of a whole organism. Since it is widely recognized that the mouse is the best animal model for human disease and development, there has recently been a tremendous surge in mutagenesis efforts around the world. A fusion of technologiesClearly, the most difficult future task is to utilize the vast amount of physical information that has been generated by the Genome Project to dissect genome function. This requires an understanding of all the 75,000 genes or so in our bodies. Generally, analysis of null mutants is the most definitive method of gene function elucidation. While targeted mutagenesis in mice has been a revolutionary technique in this regard, it will be impractical to dissect genome function on a gene-by-gene approach with standard homologous recombination technology. New, more comprehensive and global approaches are necessary. The most useful will exploit the genetic and physical maps, genomic DNA sequence, and gene expression information to move rapidly from phenotype to gene. Figure 1 depicts the course that the Genome Project has taken. High resolution genetic maps have led to the construction of genetic maps, and ESTs are being placed onto these maps. The function of the genes represented by these ESTs is the great roadblock that we face. How to best go about evaluating the function of genes in a large-scale manner? Let's look at what the world of genetics might look like a few short years from now: Tale # 1. Dr. Smith, a toxicologist, is interested in the effects of lead on brain development during embryogenesis. He decides to treat mice with lead and extract brain RNA at different points of gestation. He reverse transcribes the RNA into a radioactive cDNA probe to hybridize to a gene chip. The RNA isolation, reverse transcription and probing of the chip is done within the course of the day. The chip is then read in a device that records the expression level of 50,000 genes, revealing the marked upregulation of 18 genes. 16 are known enzymes that are not brain specific. The remaining two are known to be expressed preferentially in brain. Dr. Smith makes the conjecture that these two genes may be important for the detoxification of lead in the brain, and that lack of these genes might mimic the phenotype of lead exposure. He then purchases ES cells containing gene trap knockouts of these two genes, and injects them into mice to make mutants. This all takes place in under a week. A few months later, he will have the data. Tale #2. Dr. Jones has been performing an ENU mutagenesis screen in mice for mutations leading to muscular degeneration. She identifies such a recessive mutation, and knows it must be within a 5 cM region (about 1/300th of the genome, containing exactly 345 genes) of Chromosome 4 because the mutagenized chromosomes were screened in trans to a deletion of that region (which she obtained from a repository of mouse genome deletions she obtained from DelBank (http://lena.jax.org/~jcs/Delbank.html). Once seeing the mutant, she again checks the computer for the most current list of all skeletal muscle-expressed genes in that interval, revealing 12 strong candidates. She then exercises 3 options to nail down the mutated gene: 1) probes a gene chip with cDNA from mutant muscle, to see which of the 12 genes may be mis-expressed. 2) Orders ES cell clones from a national genome center that contain YACs known to harbor the candidate genes, to turn them into mice in an effort to rescue the mutation, and 3) Uses modern mismatch detection methods or high throughput sequencing to check each gene for point mutations. Within 2 weeks she narrows the search to a single gene that contains a splice site mutation that disrupts expression, and the transgene rescue proves the point 6 months later. These are just a couple of "science fiction" examples of the way research will be conducted in the coming years. It is already moving in that direction, with the trend being to have large centers or companies generating resources that serve an important component resource in all conceivable aspects of mammalian genomic research. Below is a brief list of existing resources for mammalian genetics research.
No doubt technologies alluded to above will be available in the coming years, including the complete genome sequence, more efficient mutation detection systems, higher density of genetic markers, banks of mice or ES cells containing YAC or BAC transgenes, efficient screening systems for mutagenized mice, and proteomics approaches for understanding interactions of gene products and dissection of biochemical or developmental pathways. Indeed the National Institutes of Health in the United States is considering all these issues at the moment. In summary, we are witnessing a coalescence of many technologies that are providing stunning access to the structure and function of the mammalian genome. These technologies are being rapidly embraced, and propelling the rate of discovery. These advances are needed, since the number of genes (about 100,000) that specify the making of a human being will take a long time to understand.
Supported by UNESCO / MIRCEN network © 1998 by Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej98006f1.jpg] |
| |||||||||

{kind=link}