 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
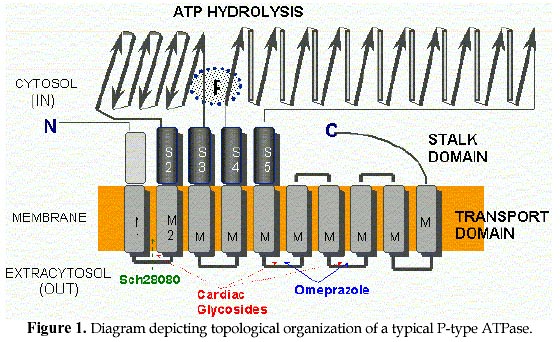
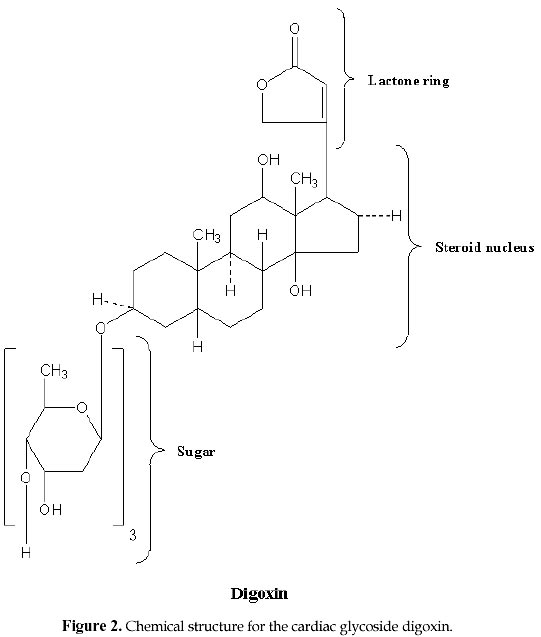
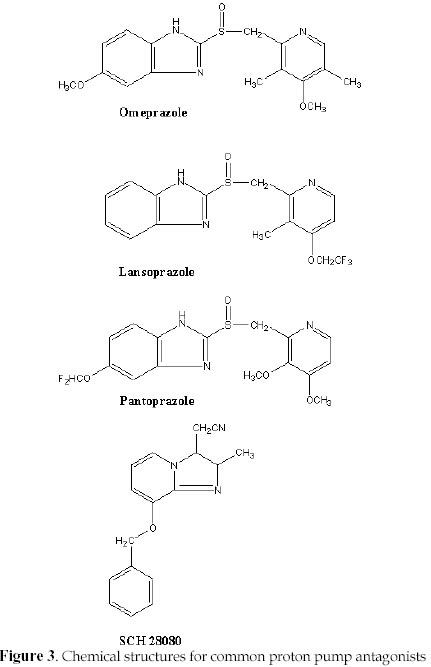
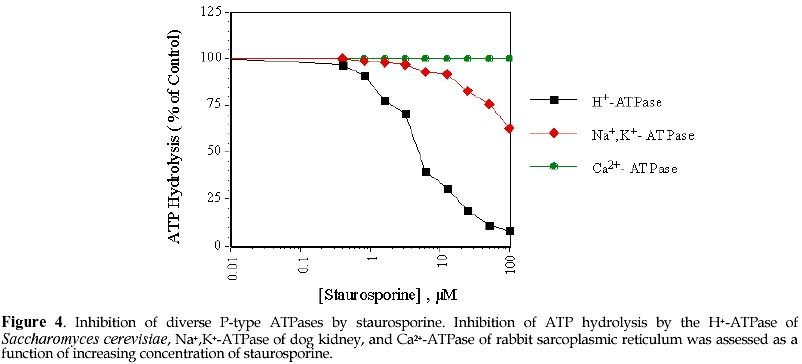
Electronic Journal of Biotechnology, Vol. 1, No. 2, August, 1998 Invited Review Article Ion pumps as targets for therapeutic intervention: Old and new paradigms David S. Perlin Public Health Research Institute, New York, NY 10016 Tel.: 212 578-0820 E-mail: perlin@phri.nyu.edu Code Number: ej98009 Abstract The development of an effective target for therapeutic intervention remains a critical part of the drug discovery process. One such target class is the P-type ion translocating ATPases, which include the Na+,K+-ATPase of cardiac cells and the H+,K+-ATPase of gastric parietal cells. These enzymes serve as selective targets for digoxin and omeprazole, which are used to treat heart disease and gastrointestinal ulcers, and are two of the leading prescribed therapeutics worldwide. It is the exquisite selectivity that can be achieved between family members that continues to make the P-type enzymes desirable targets for developing new therapeutics including a new generation of antiulcer therapeutic and a new class of antifungal therapeutic. Keywords: Ion pumps, Therapeutics, Oeprazole, Digoxin, Antifungal The modern drug discovery route starts with a disease and the identification of a therapeutic target, which can be assayed quantitatively for assessing lead compound development. Ultimately, the success of a given therapeutic will be a direct result of its effectiveness against its target, which in turn leads to an amelioration or elimination of a given disease state. Target specificity is critical to limit toxicity, although absolute specificity (one drug-one target) is difficult to achieve and even more difficult to prove. Clearly, the more that is known about the structure, function and physiological role of a specific target, the greater the chance that the drug discovery process will lead to a successful end-product. One such target class is the P-type ion translocating ATPases, which are plasma membrane-bound ion pumps involved in ionic homeostasis in all eukaryotic cells. These ion pumps include the Na+,K+-ATPase (sodium pump) of cardiac cells, which is blocked by the action of cardiac glycosides like digoxin (lanoxin) used to treat heart disease, and the H+,K+-ATPase (proton pump) of gastric parietal cells, which is selectively blocked by the action of omeprazole (prilosec), an acid blocker used to treat gastric ulcers. Lanoxin and prilosec are two of the top ten leading prescribed therapeutics world-wide, and collectively accounted for more than $3 billion in sales in 1997. Their market success reflects a positive clinical outcome for therapeutic usage. However, given the fact that the targets of these drugs are very closely related, it is the specificity of the drug-target interaction that ensures clinical outcome. This brief review will focus on the molecular properties of P-type ion pumps that make them successful targets for existing therapeutics, and will discuss how these properties are being exploited for the development of new classes of therapeutics directed against pathogenic fungi. Overview of P-type ion pumpsThe P-type ion-translocating ATPases comprise a large family of three subtypes consisting of more than 50 membrane proteins in animal, plant, bacterial and fungal cells that are responsible for the active translocation of a variety of cations across cell membranes (Lutsenko and Kaplan, 1995). These enzymes couple ATP hydrolysis to ion transport and include the animal cell Na+,K+-ATPase, Ca2+-ATPases, H+,K+-ATPase, and Cu2+-ATPases, the lower eukaryotic H+-ATPase, Ca2+-ATPases, Na+-ATPase and Cu2+-ATPases, and the bacterial K+-ATPase, Mg2+-ATPase and Cu2+-ATPase. Their reaction cycle is characterized by the transfer of the terminal phosphate of ATP to a conserved aspartate residue to form a phosphorylated (P-) intermediate, and thus distinguishing these enzymes as "P-type". P-type ion pumps frequently play prominent roles in cell and tissue physiology, and are plentiful in eukaryotic cells. The recently completed yeast genome sequencing project illustrates the vast array of active transport systems encoded by the genome including 7 P-type pumps (Clayton et al., 1997). In animals and plants, numerous cell and tissue-specific isoforms of the Na+,K+-ATPase (Lingrel, Young and Shull, 1988; Sweadner, 1989) , Ca2+-ATPase (Guerini, 1998) and H+-ATPase (Boutry, Michelet and Goffeau, 1989; Harper et al., 1990) have been characterized and shown to possess varied transport, regulatory, and inhibitor properties. P-type pumps are distinguished from V-type and Fo-F1 type enzymes by their functional and structural properties (Pedersen and Carafoli, 1987). These latter ion pump classes are multi-subunit complexes of more than 9 different subunits that are largely proton pumps involved in either ATP synthesis or intracellular acidification (Boyer, 1997; Forgac, 1992). In contrast, the P-type pumps are involved solely in ionic homeostasis and all members have a large catalytic subunit (a -subunit), usually ~100 kDa, that performs the ATP hydrolysis and coupled ion transport reactions. In some specialized animal cell enzymes, such as the Na+,K+-ATPase and H+,K+-ATPase, an accessory glycoprotein subunit (b ) of approximately 35 kDa is required for proper assembly and catalytic activity (McDonough, Geering and Farley, 1990). The enzymes are believed to function solely as monomers, although dimeric or multimeric complexes may be important under some circumstances. At the structural level, P-type enzymes share a common membrane organizational motif, which is shown diagrammatically for the P-2 subclass (Lutsenko and Kaplan, 1995) of enzymes in Fig. 1. The enzymes generically consist of a membrane transport domain with 10 transmembrane segments, a large cytoplasmic ATP hydrolysis domain, and a narrow stalk domain which links the two larger domains. It has been estimated that less than 10% of the enzyme mass is exposed at the extracellular face of the membrane, while a majority of the mass is located in the cytoplasm. A distorted mushroom-like form for the enzyme mass is supported by recent cryoelectron microscopy studies at the 8 Å level for both the Ca2+-ATPase and H+-ATPase (Auer, Scarborough and Kuhlbrandt, 1998; Zhang et al., 1998). Structural conservation is strongly maintained for P-type enzymes, even when overall primary amino acid sequence homologies are as low as 25%, as is the case for the fungal H+-ATPase and the SERCA1 calcium pumps of sarcoplasmic reticulum. Generally, the primary amino acid sequence varies considerably (25-60%) between enzyme types with different ion specificities (e.g. Na+,K+-ATPase, Ca2+-ATPase, and H+-ATPase), while it is usually conserved at high levels (>80%) within a transport type (e.g. H+-ATPases) (Wach, Schlesser and Goffeau, 1992). However, all P-type enzymes maintain certain conserved sequences and sequence motifs in the catalytic and membrane sectors that are essential to function (Lutsenko and Kaplan, 1995). A significant amount of sequence diversity occurs in the extracellular turn portions that link transmembrane segments (Monk and Perlin, 1994). These regions are known to influence binding of antagonists like the cardiac glycoside ouabain with the Na+,K+-ATPase and most likely account for the high degree of reactivity of omeprazole with the H+,K+-ATPase. It is the structural conservation that helps define P-type enzymes and enables them to behave mechanistically in a similar manner. Yet, it is the amino acid diversity between different members of the P-type class that accounts for differences in ion selectivity, regulation, and inhibitor sensitivity. It is this embedded diversity within a standardized organized structure that facilitates pharmacological approaches to these enzymes. Cardiac glycosides and the sodium pumpCongestive heart failure remains one of the most common causes of death and disability in industrialized countries, accounting for several hundred thousand deaths each year. Pharmacological intervention at an early stage of clinical disease is essential to prolong life, although mortality rates for 5 year survival still approach 50 % (Kelly and Smith, 1996). Heart failure may result from systolic and/or diastolic ventricular dysfunction, and is most commonly observed in individuals with advanced atherosclerosis. Systolic dysfunction due to idiopathic or ischemic cardiomyopathies is characterized by large, dilated ventricular chambers, while diastolic dysfunction due to hypertension, stenotic valve disease or primary hypertrophic cardiomyopathy leads to thickened, poorly compliant ventricular walls (Kelly and Smith, 1996). Therapeutic regimens must be tailored to suit individual conditions, and should take into account potential reversibility of certain myocardial deficiencies. For centuries, dating back to the description of the foxglove plant, Digitalis purpurea, in 1785 for therapeutic value, digitalis and other related cardiac glycosides have been used to treat congestive heart failure. Cardiac glycosides have a positive inotropic effect on heart output by increasing the velocity of shortening of cardiac muscle, which generates increased stroke work for a given filling volume. The increased cardiac output ameliorates the disturbances characteristic of heart failure including venous congestion, edema, dyspnea, orthopnea and cardiac asthma (Kelly and Smith, 1996). The molecular basis for the increased force of contraction is largely due to an increase in cytosolic Ca2+ during systole that increases the velocity and extent of muscle shortening. The rise in Ca2+ concentration is an indirect result of inhibition of the Na+-pump in the sarcolemma membrane by cardiac glycosides, which leads to an increased concentration of cytosolic Na+ and a diminished Na+ gradient across the membrane. The decreased Na+ potential leads to a reduced driving force for the high capacity Na+-Ca2+ exchanger, which helps regulate Ca2+ stores. A diminished Ca2+-efflux leads to a higher steady-state level of Ca2+ that can be used by contractile elements during cell depolarization cycle. Careful patient dosing leading to controlled inhibition of sodium pump activity is critical to avoid toxicity as small increases in sodium concentration can have pronounced effects on subsequent Ca2+ availability. The toxicity associated with cardiac glycosides may actually result from excessive intracellular Ca2+ that causes a transient late depolarization followed by an after contraction . Cardiac glycosides are divided into two main types of steroids: the C24 bufadienolides and the C23 cardenolides (Fig. 2). The glycosides are comprised of a sugar and a cardenolide. It is the cardenolides class that is most common and includes the digitalis analogs. Cardenolides are classified according to the chemical composition of their aglycones as lanataglucosides A-E. The woolly foxglove plant, Digitalis lanata, contains all five derivatives accounting for its severe toxicity to animals. Digoxin (lanoxin) and digitoxin, are the most commonly used digitalis members and differ from each other by the absence of a hydroxyl group at position C12 on the digitoxin molecule. Digoxin has more favorable pharmacokinetics showing rapid bioavailability from oral administration and a relatively long half-life of 14-60 h for elimination through the renal route. Inhibition of the Na+,K+-ATPase by cardiac glycosides occurs by binding to a specific site on the extracytoplasmic surface of the catalytic a -subunit. Cardiac glycoside affinity for the Na+,K+-ATPase over other animal cell P-type enzymes including the Ca2+-ATPase of sacroplasmic reticulum and H+,K+-ATPase of gastric mucosa can exceed 10,000 fold, depending on the isoform of the Na+,K+-ATPase. Enzyme isoforms from different tissues show a wide range of sensitivity to ouabain with the cardiac enzyme being approximately 100-fold more sensitive than enzyme from red blood cells. Ouabain binds in a reversible manner to the a -subunit of the Na+,K+-ATPase following phosphorylation by ATP, which yields a characteristic E2P conformational state. In the presence of K+, which promotes dephosphorylation of the E2P complex and a change in enzyme conformation, the affinity of the enzyme for ouabain is greatly diminished (Hansen, 1984). Binding to the a -subunit requires the presence of an unsaturated lactone ring at C17, a b -hydroxyl group at C 14 and a cis fusion of the A-B and C-D rings of the steroid core molecule (Hansen, 1984; O'brien, Wallick and Lingrel, 1993). The glycoside moiety does not significantly affect binding affinity but does alter pharmacokinetic properties . The molecular recognition site on the extracellular portion of the enzyme is complex and appears to require N- and C-terminal portions of the membrane domain. Early clues to the nature of binding were obtained by examining species-specific differences between ouabain-sensitive and relative resistant isoforms of the Na+,K+-ATPase from sheep and rat, respectively. Residues Gln111 and Asn122 in transmembrane segments 1 (TM1) and the extracellular turn region linking TM1 and TM2 were found to account for affinity differences (Price and Lingrel, 1988). More detailed genetic studies utilizing site-directed mutagenesis confirmed the role of the turn region and further showed that mutations in residues Cys104 or Tyr108 transmembrane segment 1 could confer resistance. However, the binding site was not localized to the this region (Schultheis et al. 1993). Chimera experiments involving the naturally resistant Ca2+-ATPase and H+,K+-ATPase showed that these enzyme acquired ouabain sensitivity when N- and C-terminal portions of the Na+,K+-ATPase a -subunit were used to substitute for comparable portions of these enzymes. These findings were supported by studies showing that ouabain affinity is influenced by residues Phe786 , Leu793, Thr797, in the extracellular loop linking TM5 and TM6, and Phe863 and Arg880 in the TM7 and TM8 hairpin. Thus, a complex binding region has emerged that involves TM1/TM2, TM5/TM6 and TM7/8. Cardiac glycoside binding in the C-terminal portion of the enzyme is intriguing because recent studies have suggested that TM5-TM8 contain the sites for ion binding and likely contribute critical features of the ion translocation domain. The well studied ion binding affinity changes on the extracellular portion of the Na+,K+-ATPase during catalysis of ATP would suggest affinity changes for cardiac glycosides at different stages of catalysis if compounds like ouabain utilize a portion of the ion translocation domain for tight binding (Lingrel et al. 1997). Finally, it is clear that the pronounced affinity of cardiac glycosides for the Na+,K+-ATPase over related animal cell P-type enzymes resides in subtle differences in the structural properties comprising the extracellular portions of the enzymes. This diversity facilitates specific interactions that confer enzyme selectivity. Finally, the highly conformationally-coupled properties of the P-type enzymes, which extends from the cytoplasmic catalytic sector across the bilayer to the extracytoplasmic domains, enable molecules bound to key extracellular regions to be effective antagonists of enzyme action. Extracellular protein diversity and transmembrane coupling are themes that will be repeated for the antiulcer acid blockers of the H+,K+-ATPase and are being exploited for the development of antifungal drugs to the H+-ATPase. Ulcers and acid blockersExcessive gastric acid production is closely linked to a number of disease states including duodenal and gastric ulcers, Zollinger-Ellison Syndrome, Gastroesophageal Reflux disease, and an assortment of other conditions. About 20 million Americans develop at least one ulcer during their lifetime, and each year ulcers affect about 4 million people. More than 40,000 people have surgery because of persistent symptoms or problems from ulcers, and about 6,000 people die of ulcer-related complications. Peptic ulcers generally arise due to an imbalance of acid secretory mechanisms and mucosal protective factors. Type I ulcers, which typically occur higher in the stomach frequently result from impaired mucosal protective factors and are not linked to hyper acid secretion. In contrast, Type II ulcers gastric and duodenal ulcers are associated with elevated acid secretion. The etiology of gastric and duodenal ulcers is complex, but it is closely correlated with infection by Helicobacter pylori, malignancy (less likely for duodenal ulcers), and use of nonsteroid anti-inflammatory drugs like aspirin. Mucosal colonization by Helicobacter pylori contributes to defects in mucosal defenses, and the eradication of the bacterium is an essential part of antiulcer therapy. It is estimated that global Helicobacter pylori infection rates vary between 20-90% and are linked to age and socio-economic status. The major pathways regulating parietal acid secretion include neural stimulation via the vagus nerve, endocrine stimulation via gastrin release, and paracrine stimulation by local release of histamine. Vagal stimulation and gastrin release stimulate the release of histamine from masts cells that activates parietal H2 receptors, which are linked to the stimulation of adenyl cyclase. Activation of the cyclic AMP pathway stimulates the H+,K+-ATPase on parietal cells, a high capacity proton pump, with its insertion into the apical membrane leading to the formation of a secretory canaliculi (Brunton, 1996). Acid secretion leads to an H+ concentration of 0.1 N in the gastric lumen. Therapeutic regimens intended to reduce acid secretion have exploited numerous aspects of this pathway. For example, the H2 receptor antagonists in clinical use such as cimetidine and ranitidine competitively inhibit the interaction of histamine with H2 receptors. In recent years, antagonists of the final mediator of acid secretion, the H+,K+-ATPase, have become the preferred therapeutic of choice due to their high degree of target specificity and clinical efficacy. The proton pump inhibitors omeprazole (losec), lansoprazole (prevacid), and pantoprazole belong to the chemical family of substituted benzimidazoles (Fig. 3), and exist as pro-drugs that need activation for interaction with the gastric H+,K+-ATPase (Lorentzon et al., 1997). The compounds are chemically stable weak bases at neutral pH, and can be orally administered. The stable pro-drugs reach the parietal cells from the blood and are transported into the strongly acidic environment of the secretory canaliculi. Protonation of these compounds results in extensive molecular rearrangement followed by the formation of a positively charged, sulfenamide species that has low permeability coefficient across membranes and reacts covalently with free sulfhydryl group(s) on the extracellular surface of the H+,K+-ATPase (Sachs et al., 1995). Significant conversion of the pro-drug to the reactive sulfenamide species requires a pH<4.0 (pKa omeprazole ~ 4.0) with more than 99.9% of the drug being rapidly converted at pH 1-2 (Brandstrom et al., 1989a). Unreacted sulfenamide is rapidly hydrolyzed in the aqueous compartment (Brandstrom et al., 1989a). The sulfenamide forms a disulfide that covalently modifies the enzyme from its extracellular domain and inhibits it with a stoichiometry of approximately 2 molecules of drug per a -subunit of the H+,K+-ATPase. Inhibition by omeprazole is believed to result from covalent modification of Cys813 (or Cys822) and Cys892 located in the extracellular loop regions joining TM5/TM6 and TM7/TM8 (Fig. 1) (Lorentzon et al., 1997;Shin et al., 1997). These transmembrane segments have been implicated in ion transport by P-type enzymes suggesting that enzyme inhibition may result from a defect in ion translocation. Such a defect would be expected to have a pronounced affect on ATP hydrolysis by the enzyme as is observed, since ion translocation and ATP hydrolysis are tightly coupled in these enzymes. Enzyme inhibition can be reversed in vitro by the addition of reducing agents such as b -mercaptoethanol, but it is essentially irreversible under physiological conditions. New acid secretion will only result from biosynthesis and assembly of new enzyme, since the turnover rate of the enzyme is ~50h (Sachs et al., 1995). Thus, the antagonistic affects of the drugs persist long after plasma clearance, which helps account for their therapeutic efficacy. Unlike classical enzyme inhibitors, which derive their specificity from lock and key-type structural mechanisms, the highly selective nature of covalent proton pump antagonists is derived from their accumulation in the acid environment of the secretory canaliculus, which activates pro-drug in close proximity to its target. In principle, the highly reactive sulfenamide species generated can react with any protein. But its accessibility to a reactive moiety on the H+,K+-ATPase, and its short half-life in the aqueous environment (Brandstrom et al., 1989b) confer exquisite specificity to these drugs. One consequence of the long-lasting action of covalent gastric H+,K+-ATPase antagonists is an elevated level of circulating gastrin that can exert a tropic effect on the gastric mucosa. The first developmental antiulcer proton pump inhibitors of shorter acting duration were the imidazo[1,2-a]pyridines and imidazol[1,2-a]-pyrazines like SCH 28080 (Brown et al., 1990) (Fig. 3). These compounds are freely reversible K+ competitive inhibitors (unlike their covalent cousins which require reducing agents to restore activity) of the H+,K+-ATPase that block acid secretion with an IC50~13 nM (Brown et al., 1990). SCH 28080 inhibits K+-stimulated ATP hydrolysis with a Ki~20-50 nM by forming a complex with the phosphorylated (E2-P) form of the enzyme in which the drug competes with K+ to prevent K+-stimulated dephosphorylation of the enzyme (Shin et al., 1998). The imadazopyridines inhibit the H+,K+-ATPase from the extracellular surface by interacting with the extracellular portion of M1 and the turn region joining M1/M2 (Lyu and Farley, 1997;Munson et al., 1994). This region of P-Type enzymes does not appear to play a direct role ion binding, but it has been shown to be conformationally-linked to the catalytic ATP hydrolysis region (Monk et al., 1994;Perlin and Haber, 1998). Compounds like SCH 28080 exhibit a high degree of target selectivity with inhibition of the gastric H+,K+-ATPase exceeding that of related P-type pumps by many orders of magnitude. It has been postulated that this high degree of specificity can be accounted for by amino acid diversity in a 12 amino acid stretch (V115AAAICLIAFAI126) in the C-terminal part of M1 and in the loop region linking M1 and M2 (Lyu and Farley, 1997;Monk et al., 1994;Monk and Perlin, 1994). The turn region appears highly conformationally active and is predicted to be as short as 4-5 amino acids in the yeast H+-ATPase and as many as 25 amino acids in SERCA1 (Monk et al., 1994). Other reversible H+,K+-ATPase inhibitors are available that are structurally dissimilar to the imidazopyridines like the acyl-quinoline SKandF 96067 and appear to inhibit the enzyme by a similar mechanism (Pope and Sachs, 1992). The reversible inhibitors are all weak bases that accumulate in acidic compartments that further adds to the selectivity of the compounds by concentrating the compounds near their site of action. These compounds are also accumulated in the acidi spaces of lysosomes and golgi produced by the action of the complex multisubunit V-type proton pump (Forgac, 1992). However, these compounds are largely inactive on this class of ion transport enzyme (Pope and Sachs, 1992). The potential of reversible inhibitors of the H+,K+-ATPase for antiulcer therapy is enormous as their potent enzyme inhibition properties can be regulated by the pharmacodynamic properties of the compounds rather than by synthesis and assembly of new proton pumps. As these various compounds progress through clinical trials, their value as therapeutics and potential to supercede their highly successful covalent counterparts will directly relate to potent and selective inhibition of the gastric H+,K+-ATPase. Developing new antifungals to the proton pumpOpportunistic fungal infections are frequent complications of HIV-infected and other immunocompromised patients. Candida albicans is the principal pathogen responsible for such infections, although other pathogens such as Cryptococcus neoformans, Aspergillus fumigatus, Histoplasma capsulatum, Pneumocystis carinii and other Candida species are important as well (Dixon et al., 1996;Meunier, 1996;Rinaldi, 1996). These organisms have become major nosocomial causes of morbidity and mortality in the immunocompromised (Crislip and Edwards, 1989;Sternberg, 1994). Candida albicans is normally found on mucosal surfaces of the mouth, gastrointestinal tract and female reproductive system. It is usually contained through competition with other commensal organisms, and the action of host defense systems such as an intact epithelium, salivary secretions, and antibody and cell-mediated immunity (Odds, 1987). Suppression of these systems by antibiotic therapy and or by disease provides a competitive advantage that allows Candida albicans to dominate the topical flora. Mucosal Candida infections are frequently observed in AIDS and other immuno-compromised patients as oral and esophageal thrush (Rinaldi, 1996), and as vaginitis in women, which accounts for more than 10 million cases per year. Mucosal infections can be effectively treated with existing polyene- and azole-based antifungal agents such as nystatin, miconazole, fluconazole, etc. In contrast, disseminated or invasive fungal disease is a more serious condition that is commonly observed in severely neutropenic patients resulting from cancer chemotherapy and organ or bone marrow transplant therapy. The percentage of cancer patients with evidence of an invasive fungal infection ranges from 5-30 percent (Meunier, 1996), and fungemia now accounts for 15% of all nosocomial bloodstream infections (Wey, Motomi and Pfaller, 1988). Candida albicans is the fourth most common cause of all bloodstream infections accounting for 8% of all infections (Pfaller et al., 1998). Other organisms such as Aspergillus fumigatus frequently cause disease in cancer patients with hematologic and other malignancies. Unfortunately, mortality rates exceeding 85% are common for cancer patients with Aspergillosis. Therapeutic agents that are effective for superficial or mucosal infections are most often ineffective for use with invasive disease. The treatment options for invasive infections are extremely limited and almost always involve the use amphotericin B (Armstrong, 1993). However, recovery rates are poor and amphotericin B suffers from extreme toxicity resulting in severe side effects, including short- and long-term renal insufficiency (Hoeprich, 1992). Newer therapeutics, like fluconazole (Diflucan) and itraconazole (Sporonox) are highly successful in treating mucosal disease. But, their long term efficacy in treating disseminated disease remains uncertain due to the presence of naturally-resistant Candida species (Candida cruseii, etc.) (Rex et al., 1995) and the development of resistance in previously susceptible species (Sanglard et al., 1995). As the mortality from fungal infections grows world-wide, there is an urgent need to develop more effective therapeutics to deal with fungal disease. The present world-wide market for medical antifungals exceeds $3 billion, and It is expected that the antifungal market will experience significant increases in the next 5-10 years, as recognition of fungal infections as an important clinical problem continues to emerge. Even as the newest and most promising antifungal agents such as voriconazole and the pneumocandins progress through clinical trials and approach the market, the number of targets represented by these and existing antifungal agents remains astonishingly small. Most of the new antifungal drugs approved in the last 15 years are azole derivatives that share a common target in inhibiting the lanosterol 14-a demethylase enzyme involved in ergosterol biosynthesis. Since azole-based drugs frequently serve as substrates for multidrug-resistance pumps, they can promote drug resistance when used chronically. It is imperative that new targets be identified to expand the repertoire of antifungal options. A new mechanistic classSeveral years ago, we proposed that the plasma membrane H+-ATPase, a classic P-type ATPase, could serve as a target for a new class of antifungal drugs (Monk and Perlin, 1994;Perlin, Seto-Young and Monk, 1997). As illustrated above, the P-type ATPases serve as effective therapeutic targets for clinically important problems. The fungal plasma membrane H+-ATPase is a proton pump that helps regulate intracellular pH (Sanders et al., 1981; Vallejo and Serrano, 1989) and maintains the transmembrane electrochemical proton gradient necessary for nutrient uptake (Prasad, 1991;Serrano, 1989). It is an essential enzyme (Serrano et al., Fink, 1986) consisting of a single large polypeptide subunit of Mr ~100 kDa that represents approximately 25% percent of the total plasma membrane protein. The H+-ATPase is one of the few antifungal targets that have been demonstrated to be essential by gene disruption (Serrano et al., 1986). In addition to its role in cell growth, the H+-ATPase has been implicated in fungal pathogenicity through its affects on dimorphism, nutrient uptake, and medium acidification . The fungal proton pumps share only a modest amount of sequence identity (~30%) with P-type ATPase family members from animal cells, while they show a high degree of sequence similarity amongst diverse fungal PMA genes (Wach et al., 1992). As described earlier, the N- and C-termini, and extracellular face of the P-type ATPases show the highest divergence, which contributes to the unique catalytic and regulatory features of each enzyme. This divergence helps account for the fact that clinically active therapeutics like cardiac glycosides and reversible antiulcer acid blockers can be selectively targeted to members of the P-type class. It is this well documented therapeutic specificity that should facilitate the development of highly selective antifungal drugs. In addition, H+-ATPase antagonists, which are selective for the fungal proton pump over related animal cell ion pumps, should display broad-spectrum activity on diverse fungal enzymes because of the high degree of primary sequence similarity found amongst these enzymes. Finally, many of the important antifungal drugs in clinical use today (e.g. azoles) are limited by their fungistatic growth properties. These drugs prevent additional growth of cells but have little affect on existing cell populations. Thus, a competent (or partially competent) immune system is required to clear infections. In the case of severely immuno-compromised individuals, this clearing is not possible and large cell populations often remain as potential sources of new infection. It is preferable that antifungal agents be fungicidal and be able to kill existing cells. The H+-ATPase is an essential enzyme that is needed for both new growth and stable cell maintenance in the absence of growth. Due to its slow turnover in the membrane (~11 h), it is likely that specific inhibitors of the H+-ATPase will be fungicidal. Overall, the fungal H+-ATPase has well defined properties that facilitate drug discovery. In addition, there are a variety of high through-put screens that assess functional properties of the H+-ATPase in vitro and in whole cells (Perlin et al., 1997). Finally, the enzyme is fully amenable to detailed genetic and biochemical analyses, which facilitate an evaluation of drug-target interactions. The importance of the H+-ATPase as an antifungal target was recently demonstrated by showing that inhibition of the enzyme by the sulfhydryl-reactive reagent omeprazole was closely correlated with inhibition of fungal cell growth, and that omeprazole-induced inhibition of the proton pump was fungicidal (Monk et al., 1995). In vitro studies demonstrated that omeprazole-induce inhibition of proton transport by the enzyme was mediated via the extracellular membrane surface (Seto-Young et al., 1997). This result was consistent with genetic studies demonstrating that perturbations of extracellular protein structure regions of the H+-ATPase can attenuate the catalytic behavior of the enzyme (Na et al., 1993). It was also consistent with the known behavior of antagonists to the Na+K+-ATPase and H+,K+-ATPase, which have interaction sites at the extracellular face of the membrane and exert inhibitory effects on the catalytic ATP hydrolysis domain located within the cytoplasm (Besancon et al., 1993;Lingrel and Kuntzweiler, 1994). In the past few years, high through-put screening of natural product and combinatorial libraries have resulted in the identification of several classes of compounds that show H+-ATPase-directed antifungal properties. These compounds typically inhibit ATP hydrolysis in the 0.3-5 m M range and show potent fungicidal properties in the 0.5-6 m M range for a broad range of pathogenic fungi including Candida albicans, Cryptococcus neoformans and Aspergillus fumigatus. Target specificity is a central issue for the effectiveness of H+-ATPase antagonists. However, it is becoming clear that there is sufficient diversity between the various P-type enzyme to provide a selective window of interaction. Fig. 4 illustrates the behavior of staurosporine, a well-known protein kinase inhibitor, on inhibition of ATP hydrolysis by the fungal H+-ATPase, the Ca2+-ATPase from sarcoplasmic reticulum and the Na+,K+-ATPase from kidney. Staurosporine, which was identified from various compound library screens as possessing H+-ATPase-directed antifungal activity, inhibits the the H+-ATPase with an IC50~5 m M. In contrast, the Na+,K+-ATPase is weakly inhibited at 100 m M and the Ca2+-ATPase is unaffected at this concentration. It is not likely that staurosporine will be an effective antifungal agent because of its broad spectrum inhibitory properties. But it illustrates the type of specificity that can be expected from large-scale compound screening against the H+-ATPase, which bodes well for the development of new antifungal compounds. ConclusionThe search for new therapeutic targets remains a primary goal of the pharmaceutical and biotechnology industries, and a great deal of emphasis has been placed on innovative approaches for target development such as those emerging from large-scale sequencing of genomes, DNA chip-based gene expression systems, and high resolution structure analyses. However, certain existing targets offer promise for future therapeutic development because drug selectivity can be achieved between like family members. The cardiac Na+,K+-ATPase and gastric H+,K+-ATPase are P-type ion translocating ATPases that serve as targets for two of the most clinically successful therapeutics, digoxin and omeprazole, respectively. These therapeutics, which are used to treat heart disease and gastrointestinal ulcers, exhibit a high degree of enzyme selectivity that is essential to their clinical success. The H+,K+-ATPase remains a target for a new generation of non-covalent proton pump antagonists, and the fungal plasma membrane H+-ATPase has emerged as an important new developmental target for the identification of a new class of antifungal therapeutic. Finally, the large number of P-type enzymes in plants, other fungi, parasites, and bacteria that remain unexploited offers promise for expanded therapeutic development. AcknowledgementsI would like to thank Mr. Steven Park for his expert assistance in drawing compound structures. The Perlin Lab is generously funded by National Institutes of Health Grant GM 38225 and by grants from Astra Hassle and Phytera. References
Supported by UNESCO / MIRCEN network © 1998 by Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej98009f1.jpg] [ej98009f2.jpg] [ej98009f4.jpg] [ej98009f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}