 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Electronic Journal of Biotechnology, Vol. 2, No. 2, August, 1999 Lipopolyamine-mediated transfection of reporter plasmids into a fish cell line Patricio Villalobos1, M. Verónica Rojas2, Pablo Conejeros3 and Sergio H. Marshall *4 1Laboratorio
de Genética Molecular, Instituto de Biología, Facultad de Ciencias Básicas y
Matemáticas, Universidad Católica de Valparaíso, Avenida Brasil 2950, Valparaíso
- Chile Fax 56 32 596703 E-mail : biaggini@ucv.cl
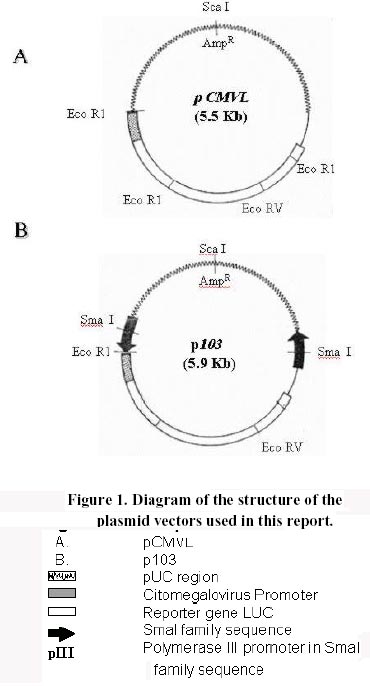
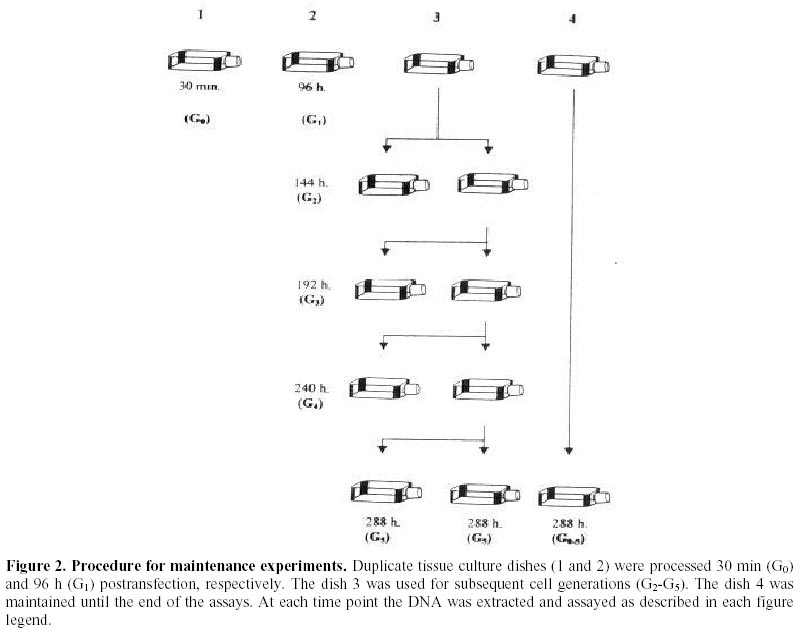
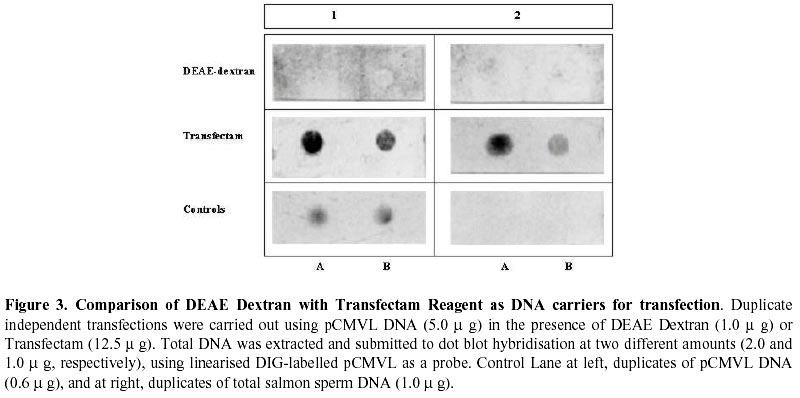
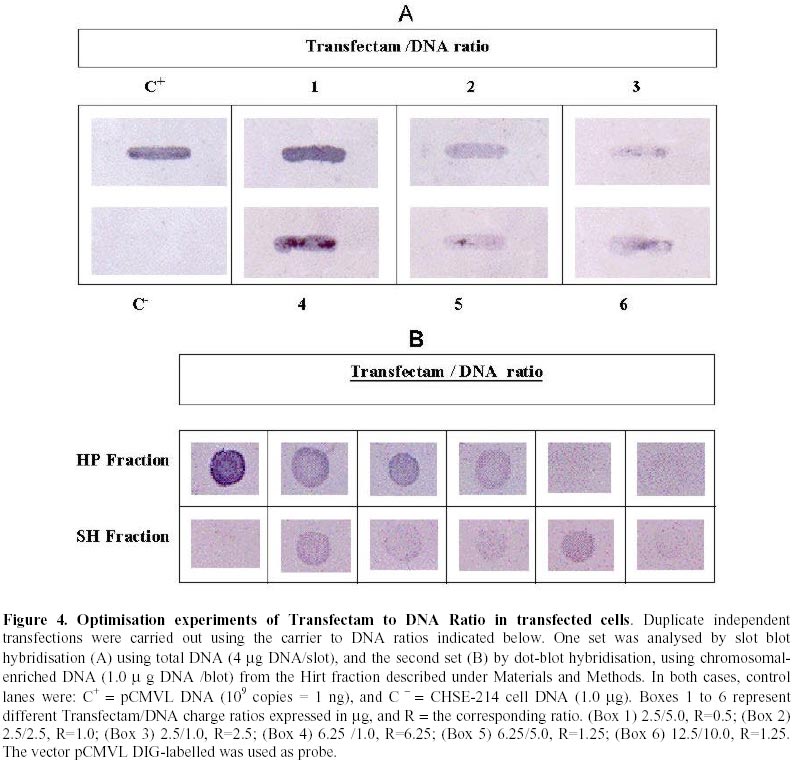
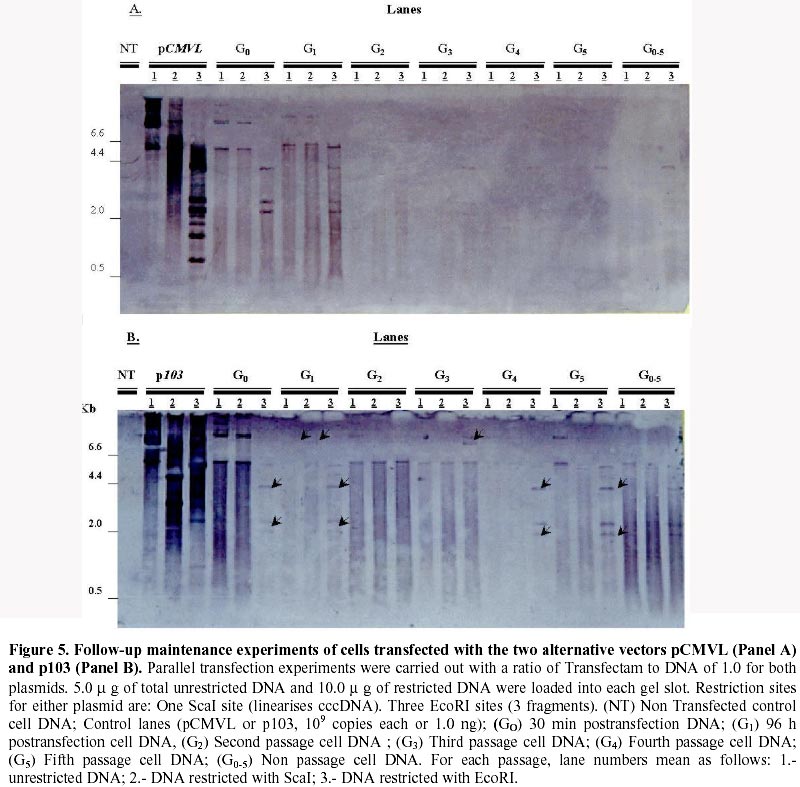
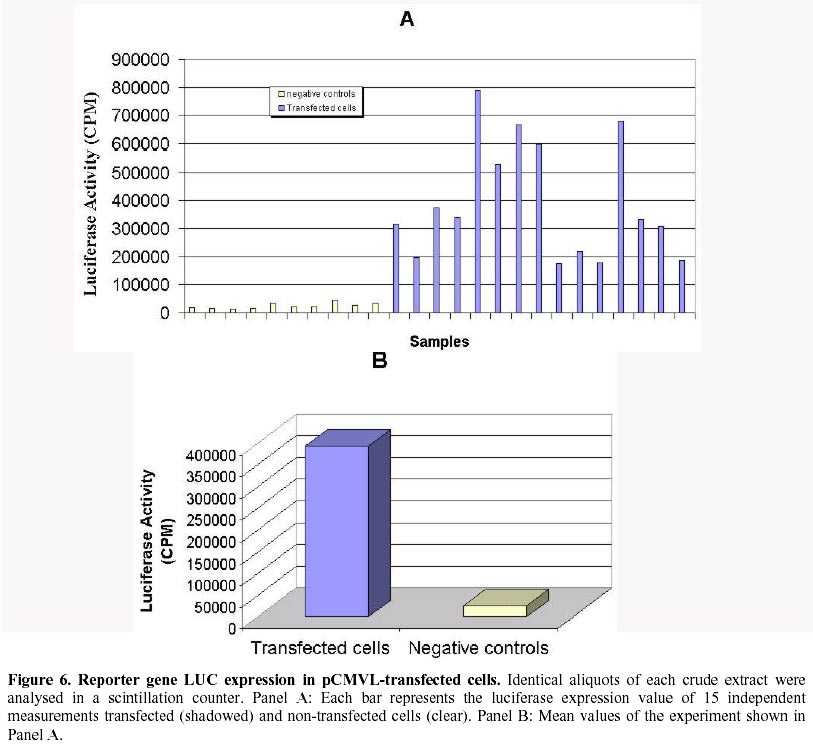
Received March 3 , 1999 Code Number: ej99012 Conditions have been optimised to transfect the fish cell line CHSE-214 to measure expression, maintenance and putative chromosomal integration of the reporter gene LUC, spliced into two versions of an expression vector. The first is pCMVL, and the second p103, a novel pCMVL-derived plasmid to which a highly conserved tandem repeat from the salmon genome was added in an inverted configuration flanking the LUC gene to promote its chromosomal integration. A minimal ratio of one to one, lipopolyamine carrier to plasmid DNA, was enough to efficiently transfect the cell line to follow the fate of target DNAs up to five cell passages. In this time-span we demonstrated the maintenance of the foreign DNA in the cells, the concomitant expression of the reporter gene, and a higher stability of p103 over the control plasmid which might suggest a higher potential for integration. Thus, we define an efficient model system for future in vitro evaluation of potential target genes of commercial interest for fish transgenesis. Keywords : Chromosomal integration , DNA maintenance , Fish cell line , In vitro transfection , Reporter gene expression. The production of transgenic animals is now established as a powerful technique that can be utilised for basic and applied research (Clark et al., 1992). The range of species which have been made transgenic has extended to fish because of an obvious potential economic benefit to aquaculture (Maclean and Penman, 1990; Maclean and Rahman, 1995; Hackett, 1996; Iyengar et al., 1996). Notwithstanding, the attempts to genetically modify fish in large scale, have been hampered by high rates of mosaicism and late integration of transgenes into the genome, which in turn result in a reduction of its vertical transmission through the germline (Penman et al., 1991; Hackett, 1993, Horvath and Orban, 1995). To date, gene transfer in fish has been achieved mainly through microinjection of individual eggs, a tedious and time-consuming technique requiring large amounts of exogenous DNA to obtain low transformation efficiencies (Yoon et al., 1990;Du et al., 1992). More recently, other investigators have produced transgenic fish by a number of alternative procedures (Araki et al., 1991), of which electroporation has given the most promising results. However, efficiencies of either integration and/or of expression of selected genes are still low (Müller et al., 1992Z, Powers et al., 1992, Sin et al., 1993; Williams et al., 1996). Lately, shotgun procedures and particle bombardment have proved to be highly efficient in delivering DNA into aquatic organisms (Cadoret et al., 1997). For fish tissues, the approach has been mostly used for vaccination purposes (Wolff et al., 1990; Kaattari and Piganelli, 1997;Robinson, 1997; Kim et al., 1997) and not yet evaluated in its potential to promote transgenesis in vivo. Fish cell tissue culture has not been properly exploited to answer key questions regarding DNA delivery and its fate in fish cells (Friedenreich and Schartl, 1990; Niels, 1991; Hernandez Betancourt et al., 1993; Köster et al., 1996). In fact, most reports available deal primarily with evaluation of regulation for gene expression (Magor et al., 1997) and only a few relate to transferring procedures (Araki et al., 1991). The most likely reason seems to be that cultured fish cells appear to be extremely sensitive to reagents commonly used to make mammalian cells competent for DNA uptake, thus restraining attempts to carry out experiments in this area. Therefore, we decided to optimise conditions for in vitro uptake of a reporter-gene containing plasmid DNA. From a number of available commercial DNA carriers, we decided to try a synthetic cationic lipopolyamine molecule. This molecule has been tested successfully in cultured eukaryotic cells with a negligible level of toxicity and without interfering with normal physiological processes (Behr et al., 1989; Murphy et al., 1998) The reagent, commercially known as Transfectam (PROMEGA USA), contains a lipid headgroup that interacts strongly with the DNA converting it into a DNA-carrier complex which mimics a cationic lipid layer. This transient structure readily associates with the target cell membrane through a cooperative ionic type interaction, which in turn promotes selective DNA uptaking and its subsequent endocytosis. Once achieved, we set a defined time-span to evaluate persistence and localisation of the foreign DNA inside the host cells, its potential rate of integration into the genome as well as the degree of expression of the reporter gene in the new environment. In order to do so, we fist had to evaluate the commercially available DNA carrier to optimise transfection conditions. Here we report the successful and efficient introduction of two related recombinant plasmids into the salmon (Onchorynchus tchawitsha) kidney embryonic cells, CHSE-214, using Transfectam, a commercial DNA carrier. After five cell passages, the foreign DNA persisted into the host cells mostly in a free form, including concatemers, both in the cytoplasm and, to a lesser extent, in the nucleus. Moreover, we also demonstrated that if the transfected DNA bear signals to theoretically promote its chromosomal integration, persistence as well as putative integration might occur by means of a non-reciprocal recombination type of event. Finally, as expected, reporter gene expression directly correlated with the maintenance of the coding sequence inside transfected cells. DNA vectors. Two related pUC-19 derived plasmids were used as expression vectors containing firefly luciferase (LUC) as a reporter gene, the cytomegalovirus early promoter, and the SV-40 polyadenylation signal to assure in vivo translation (Figure 1). The first construct is the 5.5 Kb pCMVL (Gibbs et al., 1994), and the second one is called p103 (6.0 Kb), constructed by inserting two units of the cloned Sma I family sequence (250 bp) of salmon chromosomal DNA (Kido et al., 1991) into pCMVL, flanking the LUC gene as inverted repeats (Maclean and Marshall, data not published). Based upon the evolutionary importance of the highly repetitive interspersed nature of the SmaI family in the genome of salmonids fishes (Hamada et al., 1997), the purpose of the construct was to provide a putative "transposon-like" target in the new vector, which in turn could promote chromosomal integration of the reporter gene via an homologous non-reciprocal recombination type of event. Cell transfection . CHSE-214 cells seeded to 60% subconfluency in tissue culture bottles with minimum essential medium Eagle (MEM) plus 10% fetal bovine serum (FBS), were transfected with covalently closed circular forms of the two plasmids using Transfectam (Promega, Madison, WI, USA). The media was withdrawn, and cells fed with fresh serum containing media. Transfection experiments were carried out with 1.0-5.0 m g plasmid DNA in a final volume of 500 m L of serum-deprived MEM. Either Transfectam, (2.0 m L·m g-1 DNA as specified by the manufacturer) or DEAE-dextran (100 m g·mL-1 DNA) (Holter et al., 1989) were used as DNA carriers. After 1 h. incubation at 20ºC the mixture was removed and replaced with MEM containing 10 % FBS. Cell culture passages . CHSE-214 cells were divided into two new bottles when reaching full confluency. Normally, this occurred approximately 48-96 h. after seeding old cells recovered by trypsin treatment and diluted to 50 % of the original density. This procedure was defined as "one passage". As a standard procedure, and in order to make sure that cells were fully recovered after transfection, the first passage was done after 96 h. Basically, for maintenance experiments, two sets of four 75 mL bottles were seeded each time for each vector and handled as follows: one bottle was processed 30 min post transfection, representing "generation zero" (G0). The remaining bottles were incubated for 96 h., time at which another bottle was processed representing "generation one" (G1). The third bottle was trypsinized, diluted and divided into two new ones. After 48 h., one bottle was processed as "generation two" (G2) and the other used as a source for new generations (G3 – G5). The remaining fourth original bottle was maintained until the end of the experiment and processed as (G0 - 5) at the time G5 was processed. Figure 2 summarises the experimental protocol described. Cell fractionation and total DNA purification . Cells were fractionated and DNA recovered from cells harvested at different times after transfection. For total DNA, the SDS-Phenol-CHCl3 standard procedure was used (Sambrook et al., 1989). When further fractionation was desired, the method of Hirt, (1967) modified by Panganiban and Temin (1983; 1984) was used. The procedure is based on a differential salt extraction of high molecular weight DNA from "soluble" or low molecular weight. This results in two different fractions: one soluble, enriched with free plasmid DNA in our case, which we define as "Hirt Supernatant" (HS), and one insoluble, containing chromosomal DNA with or without integrated plasmid DNA or "Hirt Pellet" (HP). The DNA contained in each fraction was further purified by successive phenol-chloroform extractions (Sambrook et al., 1989; Müller et al., 1993) and analysed either as one bulk fraction, or independently, as described in the corresponding figure legends. DNA characterisation. Identical aliquots of bulk DNA recovered from transfected cells were spotted onto a Hybond-N+ nylon membrane (Amersham International, Little Chalfont, Bucks, U.K.) using a dot blot manifold apparatus or transferred to the same type of membrane for Southern blot analysis following standardised procedures (Sambrook et al., 1989). All membranes were hybridised against linearised pCMVL as a probe, previously labelled with digoxigenin (DIG) under the conditions described in the DIG DNA labelling and Detection GENIUS Kit (Boehringer Mannheim, Indianapolis, IN, USA). Positive spots/bands were visualised using the anti-DIG-AP (alkaline phosphatase) conjugate revealed by colour development provided by the kit. For Southern blots, restriction enzymes used were from PROMEGA, USA. Measurement of luciferase expression. For luciferase expression, cells transfected with plasmid DNAs at a Transfectam to DNA ratio of 1 to 2, to assure DNA incorporation and subsequent expression of the LUC gene, were lysed at 30 hours post transfection following the standard procedure described by the Luciferase Assay System (PROMEGA Corp. Madison WI, USA). Basically 20 m L of cell extract were added to 100 m L of luciferase substrate immediately before submitting the sample to a quantitative scintillation counting method switched to the single photon mode (Cadoret et al., 1997). When required, lysates were kept at –80º C. Before assay, the extract was thawed and processed as described above. Selecting a suitable DNA carrier for efficient transfection assays Fish cell lines appear to be more sensitive than mammalian cells to reagents used to induce incorporation of foreign DNA (Niels, 1991; Hernandez Betancourt et al., 1993; Köster et al., 1996). We decided to compare the effect of DEAE Dextran, a normal carrier for mammalian cell transfection, with Transfectam, a gentle and commercially available lipopolyamine carrier, in their ability to induce cell DNA uptake following short cell exposure times. Figure 3 shows a comparative dot blot analysis of total pCMVL-transfected cell DNA at two DNA concentrations of duplicate experiments. After 1 h of cell exposure, Transfectam clearly displayed higher detectable levels of hybridisation than that observed using DEAE-Dextran as the DNA carrier. Therefore, this experiment besides demonstrating that Transfectam is an adequate DNA carrier to perform transfection experiments in cultured fish cells, allowed us to set the basic experimental conditions to be used in the remaining assays reported in this communication. Optimising carrier to DNA ratio for efficient transfection assays Two specific purposes were aimed by optimising carrier to DNA ratios: first, to minimise secondary effects on the surviving exposed cells induced by the carrier, and second, to decrease the extremely high DNA concentration required for each transfection assay as reported in the literature and suggested by the DNA carrier manufacturers. Figure 4 shows the optimisation of Transfectam to DNA ratio resulting from two independent experiments under six different experimental conditions. Figure 4-A shows a slot blot analysis using total cell-transfected DNA. Figure 4-B shows a dot blot equivalent for Hirt-enriched chromosomal DNA fractions. Both experiments gave comparable results. For equivalent amounts of DNA, the lesser Transfectam used the higher the amount of target DNA recovered inside the cells (3,4 and 1,5 in each figure). On the other hand, the lowest the carrier to DNA ratio, the better the incorporation of DNA achieved (1, 2, 3 in each figure). Notwithstanding, low amounts of DNA are sufficient to pick up strong detectable signals (3 in figures). For Figure 4 B, densitometric analysis confirmed these qualitative observations (data not shown). Also a ratio of 2.5/ 1 appears to be a suitable relation to perform transfection experiments in the model fish cell line CHSE-214. Persistence of plasmid DNA in transfected cells To measure plasmid persistence in time, we previously confirmed that the exogenous DNA added remained almost intact, concentration-wise, from the time of transfection to that when cells have fully achieved confluency (data not shown). Figure 5 summarises the results of parallel maintenance experiments after five cell passages of cells transfected with either plasmid. Panel A, cells transfected with pCMVL. Panel B, cells transfected with p103. In general, foreign DNA can be detected throughout the stages of both experiments. Notwithstanding, p103 consistently displayed a higher and more diverse distribution of the hybridisation signals all along, suggesting a higher stability than pCMVL. It might mean also that persistence is associated with either concatemerisation of the plasmid, or a higher rate of putative integration into the host cell genome. The logic behind this preliminary conclusion is that the unexpected signals (arrows) are preferentially seen when cell division occurs (G1 to G5). Expression of the reporter gene LUC in transfected cellsFigure 6 shows the results of 15 independent measurements of luciferase expression in pCMVL-transfected crude cell extracts. Extracts were obtained from 1 x 105 cells each 30 hours after transfection with a 1:1 ratio of Transfectam to plasmid DNA. The photon emission is achieved through oxidation of substratum in a reaction that requires ATP, Mg+ and O2. Equivalent distribution was obtained using p103 (data not shown). It is clearly demonstrated that incorporated DNA retains its full potential for target gene expression. Using a standard calibration curve constructed with triplicate dilutions of commercial luciferase, we estimated that in our assays we detect in the order of 150 molecules per cell, suggesting that on the average, most transfected cells seem to retain the target enzymatic activity. DiscussionThe reported experiments provide a suitable in vitro system to approach pending basic questions relevant to commercial applications of transgenesis in fish. These include transient expression, putative genomic integration, and mosaicism of selected target genes. Using low concentration of a lipopolyamine DNA carrier to transfect CHSE 214 cells we were able to increase cell DNA uptake, mainly due to a better cell recovery resulting from a lower toxic effects of the carrier. We decrease the concentration of Trasfectam suggested by the manufacturer (PROMEGA) five times, achieving detectable DNA incorporation both in CHSE-214 as well as in EPC cells, an alternative fish cell line routinely maintained in our laboratory (Villalobos, 1998). We were also keen to lower the extremely high number of target DNA copies per cell assumed to be required to efficiently transform cells both, in vitro and in vivo (Friedenreich and Schartl, 1990; Hernandez Betancourt, 1993). In our assays, we used as little as 1.0 m g plasmid DNA per 75 mL cell bottle. Considering that each bottle contained 2.0 - 5.0 x106 cells at the time of transfection, on the average we were exposing each cell to a range of 0.2 – 0.5 pg of foreign DNA, which for the plasmids examined here, equals 2.0 – 5.0 x 105 copies per cell, at least one log below the amount normally reported for naked DNA in such experiments (Maclean and Penman, 1990; Friedenreich and Schartl, 1990; Hernandez Betancourt et al., 1993; Köster et al., 1996). Maintenance experiments (Figure 5) touch on two important issues. The first has to do with the origin of the DNA used. From an evolutionary point of view, the enhanced hybridisation signals obtained with p103 over pCMVL indicate that the salmon-specific SmaI sequences do appear to have a role in the process. This allows us to think that the closer to the host the DNA sequences used to transfect the cells, the higher the intracellular stability of the target foreign DNA. This conclusion is supported by the fact that, nowadays, the most efficient expression vectors used for transgenic or vaccination purposes are host-cell specific (Hernandez Betancourt et al., 1993; Iyengar and Maclean, 1995; Hackett, 1996; Robinson, 1997) or slightly modified to increase its efficiency in the receptor host (Kim et al., 1997; Sebestyén et al., 1998). The second issue is that, independent of the origin, most plasmid vectors tend to persist in eukaryotic cells which also seems to be applicable to fish cells. Thus, a combination of these issues, in full harmony with the selected host, might be a reasonable strategy to design efficient vectors for transgenesis. In this study plasmid pCMVL (Gibbs et al., 1994), and its derivative p103, can be consistently detected after five successive passages, considering that each of them involves cell dilutions. This is a persuasive indication that both plasmids can replicate in a highly different cellular environment when compared to their original bacterial host. Moreover, the addition of the SmaI prototype conserved fish sequences in p103, seems to increase its survival in transfected cells, which might very well mean that a putative genomic integration of the selected sequences could be taking place. Reporter gene expression well above background was attained in cells transfected with either pCMVL or p103. Although the data presented here dealt with only one-time expression (Figures 6 A and B), additional information available in our laboratory suggests that the expression signal decreases after each cell generation, proportionally to cell doubling time (Conejeros, data not published). Although these data come from experiments done with pCMVL only, we can still correlate them with the persistence experiments discussed in Figure 5, in the sense that in our case the DNA signal also decreased after each cell generation. The relevant point regarding the expression of the reporter gene we want to emphasise is that its signal could clearly be detected in fish cells transfected with both model vectors, suggesting that its future putative replacement for a commercially useful transgene would also assure its expression in the target system. Optimising conditions for plasmid persistence and/or integration in the host genome would certainly contribute to define a potentially efficient expression vector for fish transgenesis. In conclusion, the experimental system presented offers a suitable in vitro model to evaluate potential target genes to be used in commercial fish transgenesis.
Supported by UNESCO / MIRCEN network © 1999 by Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej99012f6.jpg] [ej99012f5.jpg] [ej99012f2.jpg] [ej99012f1.jpg] [ej99012f4.jpg] [ej99012f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}