 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Electronic Journal of Biotechnology, Vol. 2, No. 3, December, 1999 Isolation by PCR-based methods of a plant antifungal polygalacturonase-inhibiting protein gene Melanie S. Arendse1, Ian A. Dubery2 and David K. Berger*3 1Biotechnology
Division, Agricultural Research Council-Roodeplaat, Vegetable and Ornamental
Plant Institute, Private Bag X293, Pretoria, 0001, South Africa. E-mail : melanie@vopi.agric.za
Received October 8, 1999 Code Number: ej99017
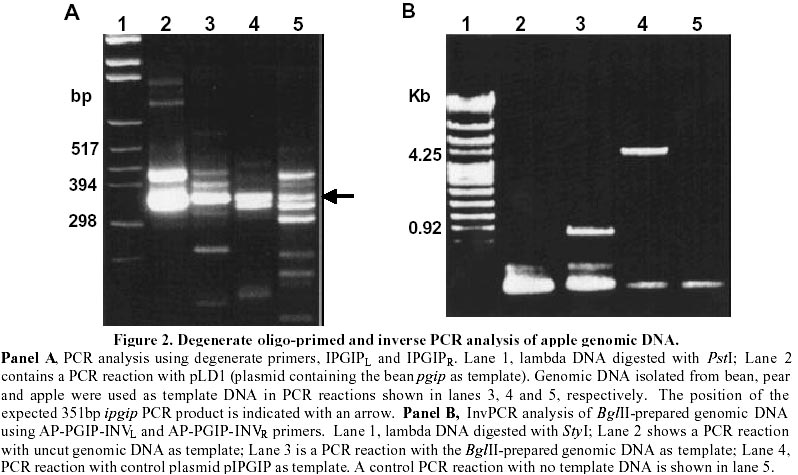
A polygalacturonase-inhibiting protein (pgip) gene from Malus domestica cv Granny Smith apple plants was cloned by degenerate oligo-primed polymerase chain reaction (PCR) and Inverse PCR. An alignment of the pear and bean PGIP sequences was used to design degenerate PCR primers in highly conserved regions. Degenerate PCR allowed the amplification of a 351bp internal fragment of the pgip gene, termed ipgip. The DNA sequence of ipgip was used to design Inverse PCR primers. A Southern blot of apple genomic DNA probed with the ipgip fragment was used to identify restriction enzyme sites for Inverse PCR. Inverse PCR enabled cloning of the remainder of the gene, from which a composite pgip gene sequence was constructed. A new set of PCR primers were designed to the 5' and 3' ends of the gene, which allowed amplification of the full-length gene from apple genomic DNA. This method has broad application to isolation of homologues of any gene for which some sequence information is known. Diseases resulting from pre- and post-harvest fungal infections have an enormous deleterious impact on world agriculture (Vasil, 1998). The recent rapid advances in agricultural biotechnology suggest that genetic engineering for increased quantitative resistance will make an increasing contribution to future disease control. Genes identified as encoding anti-microbial proteins can now be rapidly and precisely introduced into elite germplasm, creating novel pathogen resistant lines (Shah, 1997). An important part of the strategies designed to engineer increased resistance of plants to fungal diseases is the discovery and characterisation of plant antifungal proteins and the isolation of their encoding genes. The polygalacturonase-inhibiting proteins (PGIPs) are key members of a class of anti-fungal proteins that potently inhibit the activity of cell wall-degrading fungal enzymes (De Lorenzo and Cervone, 1997). PGIPs form high-affinity complexes with fungal endopolygalacturonases, thereby modulating the catalytic activity of the endopolygalacturonases. This favours the accumulation of elicitor-active oligogalacturonides that in turn trigger other defence responses. Conventional PCR allows the amplification of sequences within known boundaries. Several methods have been developed for the amplification of DNA sequences that flank regions of known sequences. These include TGW-PCR (targeted gene walking PCR) (Parker et al., 1991), UP-PCR (unpredictably primed PCR) (Dominguez and Lopez-Larrea, 1994) and Inverse PCR (Triglia et al., 1988; Ochman et al., 1988; Silver and Keerikatte, 1989). Inverse PCR allows the amplification of sequences that lie outside the boundaries of known sequences by inverting the unknown sequence. This is done by self-ligating digested DNA and opening the circular DNA molecules at a different site. This paper describes the cloning of an apple pgip gene from Malus domestica cv Granny Smith by degenerate oligo-primed PCR and Inverse PCR. The advantages and drawbacks of the PCR techniques employed are discussed. Materials and MethodsDNA isolation Genomic. DNA was obtained from young Granny Smith apple leaves using the method from Murray and Thompson (1980). Plasmid DNA was isolated with the Qiagen-Tip100 plasmid purification kit (Qiagen, Chatsworth, USA.) Nucleotide sequencing. Double-stranded DNA was subjected to automated sequencing in a Pharmacia Biotech AB Automated ALFexpress DNA sequencer using a Thermo sequence fluorescent labelled primer cycle sequencing kit (Amersham International, Little Chalfont, U.K.). Sequence analysis and alignments were done using the computer software Genepro Version 6.10 (Riverside Scientific Enterprises, WA, USA) Southern Blot Analysis. Aliquots of 10mg apple genomic DNA were digested with restriction enzymes EcoRI, BamHI, BglII, HindIII, BclI, XbaI, NsiI and SacI, respectively. The DNA was transferred onto MSI membrane (Micron Separations Inc., Westborough, USA) (Southern, 1975) and probed with a digoxygenin-11-dUTP-labelled ipgip fragment. Chemiluminescent detection, using CDP-Star ( Roche Diagnostics-GmbH, Germany) as substrate, was used. Degenerate PCR. Two degenerate primers, IPGIPL and IPGIPR, were designed to conserved internal regions between the bean (amino acids 124-129 and 235-240) (Toubart et al., 1992) and pear (amino acids 113-118 and 224-229) (Stotz et al., 1993) PGIPs (Fig. 1(a) and 1(b)). PCR was performed in 0.2ml thin-walled tubes in a MJ Research Minicycler with an internal temperature probe. Standard PCR reactions were carried out in 10ml vols. with the following final concentrations: 50mM KCl, 10mM Tris-HCl (pH9.0 at 25°C), 0.1% Triton X-100, 1.5mM MgCl2, 100mM each of dATP, dCTP, dGTP, dTTP, 0.5mM of each primer, 0.5U Promega Taq DNA polymerase (Promega, Madison, WI, USA). Standard amounts of DNA were added: 30ng plant genomic DNA or 1ng plasmid DNA. PCR cycling conditions were as follows: an initial denaturation at 94°C for 90 sec followed by 2 cycles of 94°C for 30 sec, annealing at 37°C and extension at 72°C for 45 sec. The ramp rate between the annealing and extension steps was 3.5 sec/°C increase from 37°C to 72°C. This was followed by 34 cycles consisting of 94°C for 20 sec, 56°C for 30 sec and 72°C for 45 sec (Compton, 1990) and a final extension step of 3 min at 72°C. A 351bp ipgip PCR product was cloned into pCRII TA-vector (Invitrogen, San Diego, CA) and transformed into Escherichia coli DH5a competent cells (Chung and Miller, 1988). Positive transformants were further screened by the direct colony PCR method as set out in the pMosblue T-vector kit instruction manual (Amersham International). The 4283 bp plasmid containing the 351bp ipgip PCR product was termed pIPGIP. Preparation of DNA templates for Inverse PCR. A 40mg aliquot of apple genomic DNA was digested with either BglII or NsiI at 37°C for 16h in a total vol of 250ml. The digested DNA was extracted with an equal vol. of phenol:chloroform isoamyl alcohol (CIAA; 25:24:1) and the phases were separated by centrifugation for 5 min at 4°C in a microcentrifuge. The aqueous phase was re-extracted with CIAA (24:1). The DNA was precipitated with 1/10th vol. 3M NaAc, pH5.5 and 2.5 vols. absolute ethanol, followed by 30 min incubation at -70°C. The DNA was pelleted by centrifugation, air dried and resuspended in 50ml TE buffer (10mM Tris-HCl, 1mM EDTA, pH8.0). The DNA was self-ligated for 18h at 16°C in a 100ml reaction vol. (Sambrook et al., 1989). The ligated DNA was extracted once with phenol/chloroform followed by a CIAA extraction. The sample was ethanol precipitated as described above and resuspended in 50ml TE buffer, pH8.0. The circularised DNA was linearised by digestion with XbaI in a total vol. of 100ml at 37°C for 16h. The digested DNA was re-extracted with phenol/CIAA, precipitated and resuspended in an appropriate vol. of TE buffer, pH8.0. A 30ng aliquot of the DNA sample was used in subsequent Inverse PCR reactions. Inverse PCR. PCR was performed in 0.2ml thin-walled tubes in a MJ Research Minicycler. Standard PCR reactions were carried out in 10ml vols. with the following final concentrations: 50mM KCl, 10mM Tris-HCl (pH 9.0 at 25°C), 0.1% Triton X-100, 1.5mM MgCl2, 100mM each of dATP, dCTP, dGTP, dTTP, 0.5mM of each primer, 0.5U Promega Taq DNA polymerase (Promega) and 30ng template DNA. PCR cycle conditions were as follow: initial denaturation of 94°C for 90 sec followed by 30 cycles of 94°C for 60 sec, 58°C for 60 sec and 72°C for 150 sec, followed by a final extension at 72°C for 10 min. Cloning of the Inverse PCR fragments. Inverse PCR primers, AP-PGIP-INVL and AP-PGIP-INVR (Table 1), were designed in opposite orientation to that of normal PCR using ipgip sequencing data. An 820bp Inverse PCR product was amplified from BglII-prepared genomic DNA. Inverse PCR primers, AP-PGIP-INVL-2 and AP-PGIP-INVR-2 (Table 1), were designed using sequencing data from the BglII Inverse PCR clone. A 560bp Inverse PCR product was amplified from NsiI-prepared genomic DNA. The PCR products were cloned into the pMosblue T-Vector and transformed into Mosblue competent cells (Amersham International). The resultant plasmids were termed pAppBglII and pAppNsiI. Isolation of an internal pgip fragment from apple An internal region of a pgip gene (ipgip) from apple was cloned by degenerate oligo-primed PCR. An amino acid alignment of the bean (Toubart et al., 1992) and pear (Stotz et al., 1993) PGIP sequences was used to identify regions which were conserved, and therefore may represent important functional domains (Fig. 1a; regions A and B). The codon usage of amino acids between the bean and pear was compared manually with a universal plant codon usage table (www.kazusa.or.jp/codon/) to determine degeneracy within the primers. Amplification products obtained in PCR reactions using this primer set are presented in Fig. 2A. The degenerate oligo-primed PCR allows mismatched primers to bind at the 37°C step and elongate partially. These "extended primers" bind at the more stringent annealing temperature of 56°C resulting in PCR products of different sizes, as observed in Fig. 2A (lanes 2, 3, 4, and 5). The expected size product of 351bp was obtained for PCR reactions containing either pLD1 plasmid DNA, bean or pear genomic DNA as template (Fig. 2A, lanes 2, 3 and 4, respectively). A fragment of the same size, presumably within the pgip gene, was amplified in the PCR reaction containing apple genomic DNA as template (Fig. 2A, lane 5). This indicated the presence of pgip in the apple genome. The 351bp amplification product indicated in lane 5 (Fig. 2A), was cloned into the TA-vector, pCRII, and the resultant recombinant, pIPGIP, was sequenced. Sequence alignments between the apple ipgip region and the corresponding pear pgip region (Stotz et al., 1993) revealed a 94% identity on the DNA level and a 97% similarity on the amino acid level. Selection of restriction enzymes for Inverse PCR cloning For Inverse PCR cloning, the genomic DNA has to be restricted with restriction enzymes that flank the region of interest. A Southern blot was carried out under high stringency conditions to determine which restriction enzymes to use for the digestion of the apple genomic DNA (data not shown). Previously cloned pgip genes are approximately 1 Kb in length. BglII and NsiI, which contained hybridisation bands of this size and greater, were chosen as restriction enzymes for the digestion of apple genomic DNA for subsequent use in Inverse PCR. Isolation of the apple pgip gene by Inverse PCR Results obtained for the first step of the Inverse PCR cloning of the apple pgip gene are shown in Fig. 2B. Nucleotide sequence information obtained from the ipgip fragment enabled the design of pgip gene-specific primers, AP-PGIP-INVL and AP-PGIP-INVR (Table 1), without any degeneracy. The expected size product of 4.2 kb was obtained for the positive control reaction with pIPGIP as template (Fig. 2B, lane 4). This plasmid contains the 351 bp ipgip fragment inserted into the 3932 bp pCRII vector. The Inverse PCR primers are in opposite orientation to one another in pIPGIP, and therefore the expected product of 4,2 Kb spans the region from AP-PGIP-INVL within ipgip and around the complete vector to AP-PGIP-INVR within ipgip (Fig. 2B, lane 4). A product of 820bp was amplified in the PCR reaction containing apple genomic DNA digested with BglII (Fig. 2B, lane 3). This product was cloned to produce pAppBglII. Sequence analysis revealed that pAppBglII contained sequences that covered the regions from the AP-PGIP-INVR primer upstream to the BglII site and from the AP-PGIP-INVL primer downstream to the BglII site (Fig. 3, panel B). The pAppBglII insert shared an overlap of 98bp and 166bp with the 5' and 3'-ends of the ipgip fragment, respectively (Fig. 3). Due to the sequence identity shared between pAppBglII and the ipgip fragment, the sequences were joined using computer software. The resultant sequence contained an open reading frame (ORF) which was predicted to encode a polypeptide of 274 amino acids. The ORF was predicted to continue through the BglII site, since no stop codon was present upstream of the BglII site. On the assumption that the apple pgip gene was the same length as other cloned pgip genes, an additional 168bp downstream of the BglII site needed to be cloned in order to isolate the 3'-end of the gene. Sequence analysis of pAppBglII revealed a NsiI site upstream of the 5'-end of the pgip gene (Fig. 3, panel B). A new set of Inverse PCR primers (AP-PGIP-INVL-2 and AP-PGIP-INVR-2) resulted in the amplification of a 560bp product (data not shown). This PCR product was cloned to produce pAppNsiI. Sequence analysis showed that pAppNsiI contained 300bp downstream of the BglII restriction site (Fig. 3, panel C) which indicated a NsiI site 300bp downstream of the BglII site. A stop codon (TAA) was identified 167bp from the BglII site and this represented the 3'-end of the pgip gene. Construction of a full-length apple pgip gene Based on several observations, it was concluded that the inserts from pAppBglII, pAppNsiI and pIPGIP were from the same pgip gene. Firstly, the pAppBglII insert contained 264bp identical to the ipgip fragment. Secondly, the insert of pAppNsiI shared an overlap of 190bp with the insert of pAppBglII. Both inserts contained HindIII and EcoRV restriction sites within the identical 190bp overlap. Based on these conclusions, the DNA sequence of the 5'-end of the pgip gene (pAppBglII and ipgip sequences) was linked with the DNA sequence of the 3'-end of the pgip gene (NsiI fragment) using computer software to give the complete sequence of a composite apple pgip gene (Fig. 3, panel D). Based on the sequencing data for the composite gene, a new set of PCR primers were designed to the 5' and 3' ends of the composite gene, which allowed amplification of the full-length gene from apple genomic DNA. Sequence analysis of the full-length gene revealed that it was identical to a pgip gene from Golden Delicious apples (Yao et al., 1999; GenBank accession no. U77041) which was published during the course of this work. The use of degenerate oligo-primed PCR is a powerful method to clone new or uncharacterised genes that are related to a known gene family (Compton , 1990). Lee et al. (1988) reported on the use of degenerate primers in the cloning of the urate oxidase gene. The two most critical factors in degenerate oligo-primed PCR are the design of the primers as well as the PCR conditions. The primers should be designed to an amino acid region with minimal degeneracy in codon usage (Compton, 1990). PCR conditions must be optimised to give a balance between efficiency and specificity. Inverse PCR is a convenient and versatile method of cloning unknown sequences upstream or downstream of known sequences (Triglia et al., 1988). Inverse PCR circumvents the laborious procedures of producing and screening genomic libraries (Forster et al., 1994). It has successfully been used to isolate the seed lipoxygenase promoter from pea as well as a wound-inducible promoter from asparagus (Forster et al., 1994). Potential disadvantages of Inverse PCR have, however, also been described. One of the drawbacks of Inverse PCR is the requirement for two restriction enzyme sites that flank the priming region. The lack of data on restriction sites as well as the size of chromosomal DNA greatly reduces the successful cloning rates. An additional problem with Inverse PCR is the inefficient PCR amplification of closed circular double-stranded DNA. Forster et al. (1994) found that digestion of the self-ligated DNA improved the yields of PCR amplification product. The presence of introns in the target gene must also be considered when carrying out Inverse PCR from genomic DNA. In this study the apple pgip gene did not have introns. In the study reported in this paper, several different enzymes were used to digest the apple genomic DNA, but subsequent Inverse PCR amplification was only successful for some of the enzymes used. One of the reasons could be that the sites were too far apart so that in the subsequent PCR, the amplification of large products was inefficient. Some DNA is lost at each clean-up step after digestion, ligation and re-digestion, therefore it is difficult to determine the amount of target DNA added to the PCR reaction. The absence of amplification products could therefore be due to insufficient target template. Several PCR methods have subsequently been developed to overcome difficulties experienced with Inverse PCR. One of these methods, called random primed gene walking PCR (Trueba and Johnson, 1996) allows the amplification of specific PCR products. This enables direct sequencing of unknown regions without the need for DNA cloning. Degenerate oligo-primed PCR and Inverse PCR were used to clone a pgip gene from Granny Smith apple fruit. The pgip gene was identical to a pgip gene (GenBank accession no. U77041) reported during the course of this study (Yao et al., 1999). The deduced apple PGIP showed a high degree of similarity with previously isolated PGIPs and contained structural features characteristic of PGIPs found in several plant families. It is interesting to note that the similarities appear to fall into two distinct groups: the fruit PGIPs compared to the PGIPs from vegetables. The apple PGIP is more identical to the PGIPs from kiwi (69%), pear (97%) and tomato (64%) than to the soybean (55%) and bean (53%) PGIPs. The high degree of amino acid homology shared between the apple and pear PGIPs could be explained by the fact that both apple and pear belong to the same plant family Rosaceae, subfamily Pomoideae. The proposed structure of the apple PGIP contains 10 imperfect leucine-rich repeat (LRR) motifs that span nearly 80% of the mature peptide. Leucine-rich repeats are common motifs found in plant disease resistance (R) genes. The LRR domains in the tomato C¦ proteins have been suggested to be involved in defence signal activation by either interacting with a signalling component or by binding to a ligand (Jones and Jones, 1997). PGIPs could therefore interact with their ligands (endopolygalacturonases) by means of their leucine-rich repeat domains. To date, PGIP is the only LRR protein for which its ligand has been determined and characterised (De Lorenzo and Cervone, 1997). An understanding of the interaction between PGIP and endopolygalacturonases could reveal how plants recognise non-self molecules and convert recognition into action. References
Supported by UNESCO / MIRCEN network © 1999 by Universidad Católica de Valparaíso -- Chile The following images related to this document are available:Photo images[ej99017f2.jpg] [ej99017f1.jpg] [ej99017t1.jpg] [ej99017f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}