 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
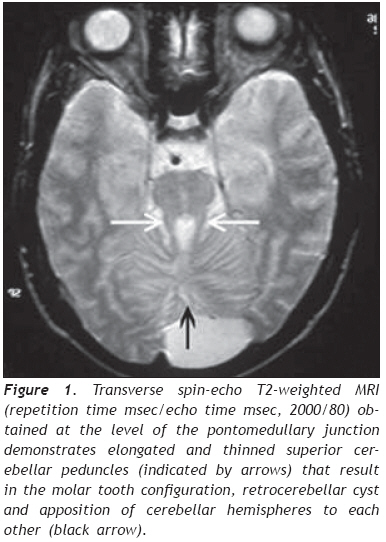
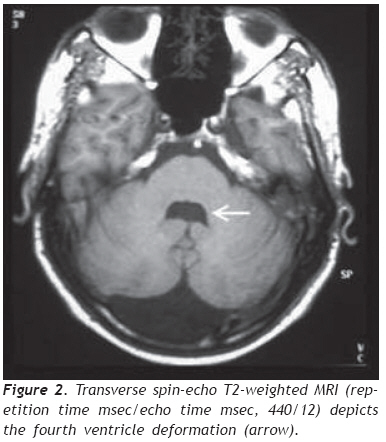
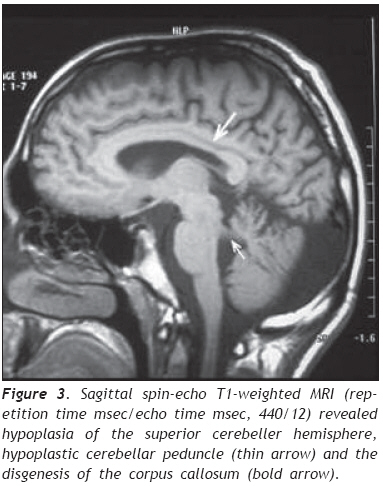
European Journal of General Medicine, Vol. 6, No. 2, 2009, pp. 119-122 A Turkish Adulthood Joubert Syndrome and Review of the Literature Mehmet Noyan Zenger1,5, Serdar Kabatas2,4, Ali Fuat Baykiz3, Yang D. Teng4 Departments of 1Radiology, 2Neurosurgery, and 3Psychiatry, Elazig Military Hospital, Elazig, Turkey 4Department of Neurosurgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, USA 5Department of Radiology, Ozel Nakiboglu Hastanesi, Konya, Turkey Code Number: gm09026 Disclaimer The authors have no financial interest in the subject presented in this paper. ABSTRACT Joubert Syndrome (JS) in adult population is extremely rare. Here, we describe the first adult case seen in the Republic of Turkey. Our patient, a 19-year-old male, was diagnosed primarily by magnetic resonance imaging (MRI) finding of the typical “molar tooth sign”, in addition to JS clinical features such as mental retardation, hypotonia, dysarthria, nystagmus and ataxia. Detailed images of high resolution MRI, a review of the literature and a brief discussion are reported. Keywords: Adult, Joubert Syndrome, MRI. INTRODUCTION Joubert Syndrome (JS) is a rare neurological disorder with agenesis or hypoplasia of the cerebellar vermis and a distorted brain stem. Primary clinical features include hypotonia, developmental delays, and either abnormal eye movements (i.e., nystagmus) or an abnormal breathing pattern (1,2). Additional deformities such as extra fingers and toes, cleft lip or palate, tongue abnormalities, and seizures may also occur. Many patients die in infancy or childhood, but some survive into adulthood with variable cognitive and motor impairments, depending on whether the cerebellar vermis is entirely absent or partially developed (3). JS is mostly presented as sporadic cases. However, there are some familial incidents which appear to be inherited via recessive genes. Here, we discuss an adult case which was diagnosed principally by magnetic resonance imaging (MRI) finding of the typical “molar tooth sign”, and by manifestation of other related clinical symptoms, including mental retardation, hypotonia, dysarthria, nystagmus and longstanding ataxia (4). To the best of our knowledge, this is the first report on an adult JS male individual in the Republic of Turkey. CASE REPORT A 19 years old male was admitted to our clinic with parental complaints on his difficulties in speech, walk and vision. His developmental history could not be followed up entirely in detail levels. It was, therefore, difficult to verify the time-course evolvement of the main complaint related problems. Physical examination showed right eye rhythmic oscillation into different directions, atrophic changes in both arms and feet, and mild hypotonia, especially in his lower extremities. The patient, however, was not found with discernable problems of breathing and other congenital anomalies. Neurological examination found rotatory nystagmus which was more definitive in the right eye, prolongation of bilateral strabismus that might be caused by bilateral nervus abducens paresis, truncal ataxia (i.e., when standing under either open or closed eye condition, his trunk gradually inclined towards the right side), mild dysarthria, and global hypereflexes (5). Ophtalmologic observations did not find retinal anomalies or degeneration. In psychiatric consulting evaluation, he was deemed as lack of appropriate awareness to surrounding environmental changes. He was found to have a mild mental retardation that was mainly judged by his Intelligence Quotient (IQ) level of 70. During the entire clinic visit his communication with physicians and staff was done with help of his father. Thus, his verbal communication appeared largely functional to people who are familiar with his life. His speech, however, was consistently slow, and includes dysarthric components. The patient showed nearly normal gait but had major difficulty in running that triggered appearance of staggering-like gait disorder, followed shortly by falling (4). He expressed through his father that he was not capable to play sports that were common for his peers. Conversely, he could perform his own personal care activities, such as independent bathing, dressing and toileting. Due to the circumstantial limits, we were not able to conduct any genetic analysis for the patient and his family. The overall clinical symptoms in addition to the main complaint of the patient indicated that this might be a very rare adult case of Joubert syndrome. We therefore focused on seeking systematical neuroradiological evidence. Transverse MRI images obtained at the level of the midbrain showed a “molar tooth sign” typical in patients with JS (Figure 1). (6). Further MRI image analysis revealed a bat wing-shaped fourth ventricle, vermian atrophy and a deep cerebellar midline cleft (Figure 2). Sagittal images additionally demonstrated hypoplasia of the superior cerebeller hemisphere, hypoplastic cerebellar peduncles and the disgenesis of the corpus callosum (Figure 3). Together the examination outcomes supported our notion, and the patient was diagnosed with JS. The patient was later discharged with his father being given general information on JB prognosis and patient support. DISCUSSION JS was first described by Dr. Marie Joubert in 1969. It has been estimated to occur in approximately 1 in 100,000 individuals and associated with retinal coloboma and dystrophy in approximately 50% of patients, and multicystic kidney disease in 30% of patients (1,2). The congenital brain malformation also comprises abnormalities of axonal decussation affecting the corticospinal tract and superior cerebellar peduncles. Thus, individuals with JS have motor and behavioral disorders, including an inability to walk due to severe clumsiness and mirror movements, and cognitive and behavioral disturbances (7). The main imaging findings, in all patients, are partial or complete absence of the vermis, hypoplastic cerebellar peduncles, and the fourth ventricular deformity. The cerebellar hemispheres are usually normal. In our case, cerebellar hemispheres are in direct contact in the midline due to severe hypoplasia of the vermis (Figure 1). The cerebrum is mostly not affected, although moderate lateral ventricular enlargement due to atrophy was described in 6%–20% of JB cases, with corpus callosum dysgenesis present in 6%–10% patients (Figure 3). (1). The term “molar tooth” refers to the characteristic appearance of an enlarged and horizontally directed tubular structure on each side of the midline emerging from the midbrain. In one earlier study, “the molar tooth sign” was identified in 85% of patients with JS, and thus was considered pathognomonic of this disorder (6). In our case, radiological evidence of JB is suggested by the combination of hypoplasia of the cerebellar peduncles that results in the molar tooth sign and severe hypoplasia of the vermis that gives a bat-wing appearance to the fourth ventricle and leads to a midline cleft between the two cerebellar hemispheres (Figure 2) (1,2). Regarding differential diagnosis, Arima and Senior-Loken syndromes with posterior fossa malformations often add difficulties in making clinical diagnosis of JS. Nevertheless, molar tooth sign, though to a less degree existing in other hindbrain/midbrain syndromes, is still considered virtually diagnostic of JS. Tectocerebellar dysplasia, rhomboencephalosynapsis, and Dandy-Walker syndrome are additional source of confusion, and they may also accompany JS (8). But in Dandy-Walker syndrome, a large cystic abnormality of the posterior fossa is seen and is often accompanied by posterior fossa enlargement. In rhombonencephalosysnapsis, unlike in JS, a midline cerebellar cleft is not present because of the cerebellar hemispheres fusion (2). Inheritance of this disease is said to be autosomal recessive. Studies have shown that it is a genetically heterogenous disorder with one locus pointing to chromosome 9q (9). More recently, Walsh and Gleeson labs studying families from Middle East identified mutations in AHI1 gene that caused the disease in these families (9,10). In JS patients with progressive kidney disease, mutation of NPHP1 gene was determined. There is a generally held perception that the genetic foundation of JS may eventually be determined to be equally as varied and heterogeneous as the clinical presentations (10,11). Thus, Saraiva and Baraitser defined diagnostic criteria (vermian hypoplasia, hypotonia, developmental delay, and either eye movement or respiratory abnormalities) are still highly valuable for diagnosing JS. In regions that lack genetic tests accessibility, neuroradiology exams should be used to assist final diagnosis (1,5,10). In the absence of general biochemical or genetic markers and animal models for the JS, currently little can be known in terms of the pathogenic mechanisms. Hence, there is no specific treatment for JS. Prognosis differs from age and associated anomalies, but is generally very poor due to the severe mental retardation in most cases. The main causes of death are severe feeding difficulties and respiratory infections with associated anomalies such as cranial meningocele (12). Respiratory abnormalities typically improve with age advancing, but cognitive and motor function typically decline (3). Treatment for JS is therefore symptomatic and supportive. Infant stimulation, physical, occupational and speech therapy may have beneficial effects (13). In conclusion, in addition to prenatal and child JB cases that were reported previously (14), we have now presented the first adult case of JS in the Republic of Turkey. Given the region novelty and age rareness of our patient, follow-up genetic investigations are highly desirable to determine if this is a case of AHI1 mutation, or perhaps a different genotype that has not been identified in Middle Eastern families. Such studies may reveal new genes that are linked with this developmental disorder. Acknowledgments We thank Dr. Dou Yu (the Spinal Cord Injury & Neural Stem Cell Biology Laboratory, BWH/HMS) for his generous help with preparation of the figures. REFERENCES
Copyright 2009 - European Journal of General Medicine The following images related to this document are available:Photo images[gm09026f3.jpg] [gm09026f2.jpg] [gm09026f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}