 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Human Genetics, Vol. 8, No. 2, Jul-Dec, 2002 pp. 52-59 Review Article p53 in Brain Tumors : Basic Science Illuminates Clinical Oncology Chitra Sarkar, Sanjay Mukhopadhyay, Mehar Chand Sharma Departments of Pathology, All India Institute of Medical Sciences, New Delhi, India.
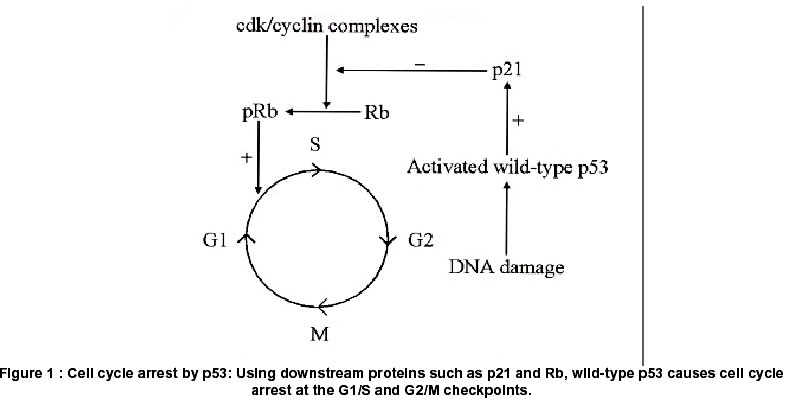
Code Number: hg02011 p53 is a tumor suppressor gene known as the "guardian of the genome" It protects cells from cancer, by preventing cells with damaged DNA from proliferating wantonly. This function is achieved by various unique mechanisms. These include cell-cycle arrest, which facilitates cell repair; and apoptosis, which ensures the death of cells too severely damaged to be repaired. We discuss these mechanisms and their significance in tumor biology. The p53 gene enjoys the dubious distinction of being the single most common target for genetic alteration in human tumors. We discuss here the role of p53 and its mutations in astrocytomas and other brain tumors. The study of p53 in astrocytomas is one of the prime examples of the close and fruitful collaboration between molecular biology and clinical practice. After decades of research, it is now accepted that p53 is involved both in the initiation as well as the progression of astrocytomas. A multi-step sequence for the evolution of astrocytomas to glioblastomas has been elucidated. The prognostic significance of p53 expression in brain tumors has also been a matter of extensive research. Finally, we touch upon the therapeutic implications of p53 in astrocytomas. Given the dismal consequences of the absence, malfunction or mutation of this gene, many workers have tried to halt the progression of tumors by reintroducing the wild-type gene as a form of biological therapy. This approach, conceptualized in the laboratory and actualized at the bedside, holds much promise for the treatment of these challenging tumors. p53: The Gene and the protein p53 is a tumor suppressor gene located on the short arm of chromosome 17 at band 13.1 and consists of 11 exons. The major role of p53 is the maintenance of genomic stability. It is therefore referred to as the "guardian of the genome" and is thought to be a critical "gatekeeper", guarding against the formation of cancers.1 There is evidence to show that p53 acts as a "molecular policeman" in preventing the propagation of genetically damaged cells. 1,2 This function is accomplished either directly by the participation of p53 in mechanisms that maintain DNA integrity, or indirectly, by the induction of cell-cycle arrest, senescence, or apoptosis of damaged cells.2 More recent data shows that p53 also has an important role in angiogenesis and tumor invasion, two processes fundamental to malignancy.2 The protein p53, derived from the p53 gene is named for its biochemical nature (phosphoprotein) and its molecular mass (53 kilo daltons). It is also called tp53, or tumor protein 53. Three independent groups, using different approaches3-5 to the study of tumorigenesis, discovered p53 in 1979. Functions of the p53 gene (i) p53 senses DNA damage and then induces growth arrest or apoptosis The p53 gene is summoned to action only when DNA is damaged. p53 functions primarily to control the transcription of other genes, the goal being to limit the proliferation of potentially cancerous cells containing damaged DNA. This involves augmentation or enhancement of the p53 gene product by a mechanism that is poorly understood. In any event, enhanced p53 acts as a transcription factor by binding to DNA. The genes that it then transcribes function to attain cell cycle arrest and/or apoptosis. (ii) p53 and cell-cycle arrest p53 suppresses the cell cycle at the G1/s and G2/m transitions. To cause cell cycle arrest, p53 recruits the phosphoprotein p21 (also called Cip1) in what has become known as the p53/mdm2/p21 pathway. p21 is a 21-kilo dalton (kda) protein encoded by the cdkn1 gene, that acts as a cdk (cyclin-dependent kinase) inhibitor.6 It inhibits a range of cdk/cyclin complexes required for cell-cycle progression. The inhibition of cdk/cyclin complexes by p21 in turn prevents the phosphorylation of the retinoblastoma (Rb) gene that normally allows cells to move from the G1 to the S phase of the cell cycle. This pause in cell cycling ("G1 arrest", the raison d'être of p53) is a welcome breather for the cell, during which the damage inflicted on DNA by mutagens is repaired.7 To achieve this repair, p53 activates the transcription of a DNA-repair protein called gadd45 (growth arrest and DNA damage-inducible).8 If gadd45 succeeds in its mission, p53 selflessly calls for its own down-regulation (by activating a gene called mdm2 or murine double minute-2)9 and cell division is allowed to proceed as before. This sequence is illustrated in figure 1. (iii) p53 and apoptosis However, if gadd45 fails in its endeavor to repair the DNA damage, p53 consigns the potentially malignant cell to its death. To this end, p53 activates the apoptosis-inducing gene bax and the death receptor Gd 95 (also known as Fas/Apo1) while inhibiting the expression of the apoptosis-inhibiting protein bcl2. Thus, p53 has a pro-apoptotic function.10 Tp53 gene mutations The p53 gene enjoys the dubious distinction of being the single most common target for genetic alteration in human tumors.11,12 This is possibly because of the diversity of the biological pathways in which p53 is involved. All classes of mutations, deletions, insertions, transitions and transversions occur in the Tp53 gene. Several mutational "hot-spots" have been observed in the p53 gene, all of which occur within four highly conserved amino acid sequences (exons 5 to 8). Although more than 250 codons in the Tp53 gene are potential human mutation sites, the most frequently observed mutations are found in only 5 of these codons-175, 245, 248, 249 and 273.13 There are two ways in which both alleles of a pair of p53 genes can be knocked out and rendered ineffectual. In most cases, both inactivating mutations occur in somatic cells of the organism. Less commonly, some individuals inherit a mutant allele in germ-line cells. All that is then needed to inactivate the normal allele is a second "hit" during the life of the individual. Such individuals are said to have the Li-Fraumeni syndrome, and have a 25-fold greater chance of developing a malignancy by 45 to 50 years of age compared with the general population.14 A wide variety of tumors, e.g. sarcomas, breast cancer, leukemia, brain tumors and adrenocortical carcinomas occur in these patients. Further, these tumors tend to occur at younger ages and are prone to multiplicity when they occur as part of the Li-Fraumeni syndrome. p53 mutations can have three consequences.15
Wild-type and Mutated p53 protein The "normal" p53 protein is called "wild-type p53", as opposed to the deranged "mutated" p53 that occurs in tumors. Under physiologic conditions, the half-life of wild-type p53 protein is short (15-30 minutes), presumably because of ubiquitin-mediated proteolysis. As a result, wild-type p53 protein is present at very low levels in normal cells16 and is undetectable by immunohistochemistry. In contrast, the half-life of mutant p53 protein is in the range of hours, resulting in high, immunohistochemically detectable levels of the protein. For all practical purposes, therefore, if the protein can be detected in tissue sections by immunohistochemistry, it is assumed that the gene is mutated.16 However, this generalization is not absolute. Thus, not every mutation of the p53 gene results in immunohistochemically detectable p53 overexpression17. By the same token, not all immunohistochemically detectable p53 protein positivity results from p53 gene mutations. Indeed, many p53 immunopositive cases are not the result of accumulation of mutant p53 protein, but rather are due to accumulation/overproduction of wild-type p53 protein. The accumulation could either be through binding of wild-type p53 to altered cellular or viral proteins or simply due to accumulation of wild-type p53 in rapidly dividing tumor cells independent of p53 mutation. Another possibility is the stabilization of wild-type p53 protein by formation of complexes.18, 19 Further, in a study on 40 astrocytomas it was found that p53 immunopositivity was governed not by alterations of the p53 gene itself but by other putative tumor suppressor genes in the chromosomal region 17p13.3.20 Similarly, tumors with p53 gene mutations but with p53 protein immunonegativity have been reported. This has been attributed to synthesis of truncated protein which either lacks or cloaks the epitopic region or prevents its binding to proteins that prolong its half-life.19 Thus, p53 immunohistochemistry may reflect several different biological conditions other than p53 gene mutation. However, immunohistochemistry for p53 protein will continue to play a significant role in characterizing the biology of this protein in neoplastic conditions because of the relative ease of the methodology compared to standard molecular techniques. p53 in brain tumors Here we review the role of p53 in brain tumor development. We will concentrate on astrocytic tumors, which are the commonest primary brain tumors, and in whom p53 plays the most significant role. The role of p53 in other tumors will be mentioned briefly. The many findings in recent years that have demonstrated the importance of the p53 gene in the development of astrocytic tumors will be highlighted. We will also discuss the potential use of wild-type p53 in the therapy of tumors in which the normal p53 pathways are disrupted or dysfunctional. p53 in astrocytic tumors According to the who,21 these tumors can be divided into four grades:
(i) The role of p53 in astrocytoma initiation/ early astrocytic tumorigenesis Evidence for causal involvement of p53 in glial tumorigenesis came from the finding that expression of exogenous wild-type p53 in glioblastoma cell lines suppressed their growth.22 In addition, tissue culture experiments conducted on the cortical astrocytes of mice indicated that loss of function of p53 resulted in the immortalization of these cells. In addition, these murine cells lacking p53 activity became aneuploid, an observation that was in keeping with the aneuploidy seen in human astrocytomas with p53 mutations.23 The notion that p53 mutations are an important factor in the genesis of brain tumors was also supported by the observation that patients with the Li-Fraumeni syndrome (see above) have a high (13%) incidence of brain tumors early in life.24 The majority of brain tumors in the Li-Fraumeni syndrome are indeed astrocytomas, in which the p53 gene is frequently mutated.25 These observations are important because mutations in the germ line (as seen in p53 mutations in malignancies of the brain, breast, bone and soft tissue) are believed to be "mutator" mutations, initiating tumor formation. Allelic loss of chromosome 17p26 and mutations of Tp53 have been observed in approximately one-third of adult astrocytic tumors of who grades ii, iii and iv, suggesting that inactivation of p53 is an early event in the formation of grade ii tumors. In concordance with the p53 gene mutation, increasing incidence of p53 protein reactivity has been reported with increasing grades of malignancy.27- 29 grade ii: 18-46%, grade iii: 29-57%, grade iv: 49-70%. Most Tp53 studies have concentrated on the conserved exons 5 to 8. However, some studies that have included all the coding sequences (exons 2 to 11) found only a handful of mutations outside exons 5 to 8. The three most commonly mutated codons reported, in order of frequency, are 273, 248 and 175. In other human tumors, the most frequently mutated codons are 248, 249 and 175. However, there are no data indicating the role of brain tumor-specific mutations.11,13,30 (ii) p53 in malignant progression of astrocytic tumors Recurrence and malignant transformation are important features of astrocytic tumors.19, 31 Two distinct types of glioblastoma are now recognized, based on the types of genetic alterations that characterize them. Primary glioblastomas These glioblastomas rarely show p53 mutations (only approximately 10% have p53 mutations) but rather have amplification of the epidermal growth factor receptor (EGFR) gene on chromosome 7, platelet-derived growth factor receptor (PDGFR) gene amplification, loss of deleted in colon carcinoma (DCC) gene expression, and loss of heterozygosity (LOH) involving chromosome 10.32 These abnormalities are typically found in older patients with a short clinical history and no evidence of progression from a lower grade astrocytoma. These tumors are hence termed primary, or de novo, glioblastomas.32 Secondary glioblastomas More than 65% of these glioblastomas show p53 mutations, of which more than 90% were already present in the first biopsy (original tumor). They occur in younger patients whose tumors have progressed from a lower grade astrocytoma over a comparatively long time period. It is possible that the genomic instability associated with p53 loss facilitates the accumulation of additional genetic mutations that promote the transformation to secondary glioblastomas. Studies that have compared pairs of primary and recurrent astrocytic tumors have found that both sets of tumors harbored similar p53 mutations. The percentage of glioma cells with p53 protein increased from the first biopsy, suggesting that the recurrence or progression occurred by clonal expansion of the p53-mutated cells in the primary tumor.33-35 Our own data show that the percentage of tumors that are p53 positive increases at recurrence, irrespective of the initial grade of the tumor. Thus, 56.25% of primary tumors were initially p53 positive, while in recurrent tumors associated with malignant progression this frequency increased to 70.83%. Further, recurrence was associated with increase in the percentage of p53 immunopositive cells. No case that was initially p53-positive in our study became negative at recurrence. In conformity with the literature, we found a higher degree of p53-positivity in secondary glioblastomas (84%) compared with de novo glioblastomas (43%)27. These genetically distinct types of glioblastoma cannot be distinguished on histological grounds alone, and it remains uncertain whether survival after diagnosis is different in these groups.32,36 p53 brain tumors To summarize, although the role of p53 in the recurrence and progression of astrocytomas is still debated, there is growing evidence34, 37-40 that: a) Genetic alterations occur with equal frequency both in low- and high-grade astrocytoma, indicating a role for p53 in the formation/initiation of low-grade disease. b) p53 also has a role in the progression from low-grade astrocytoma to secondary glioblastoma. c) p53 as a prognostic factor in astrocytic tumors p53 mutations correlate with prognosis in a number of human cancers, including cancers of the breast, stomach, colorectum and lung.41,42 In these tumors, a high rate of p53 mutation connotes increased tumor aggressiveness, a high metastatic rate and poor prognosis. The predictive role of p53 mutations in astrocytomas however is still poorly understood. Recent studies indicate that in low-grade astrocytomas that progress to glioblastoma, the frequency of p53 mutations is very high, of the order of 58-83%.28,43 Neoplasms with p53 mutations also appear to progress more frequently than neoplasms without the mutation, but evidence for this correlation remains circumstantial.44-46 The time interval to progression appears to be shorter in patients with low-grade astrocytomas that carry a p53 mutation.28 Hence, it seems that low-grade astrocytomas with p53 mutations progress to glioblastomas faster and more often than tumors without these mutations. The effect on patient survival, however, is still murky, with some studies showing that p53 mutations have no effect on clinical outcome and survival.45,47,48 Others propose that low-grade astrocytomas that carry a p53 mutation appear to be associated with a shorter survival.49 It must be noted that some of these studies are based on childhood malignant gliomas, which may be biased because p53 mutations in children occur almost exclusively in brainstem gliomas, which are known to have a very poor prognosis. On the other hand, the relatively poor prognosis of gemistocytic astrocytomas may be due to the fact that they have a higher frequency of p53 mutations. In contrast, some authors claim that the existence of p53 mutations in recurrent high-grade tumors may indicate a better prognosis.29 Our studies have shown that p53 positive anaplastic astrocytomas had significantly shorter symptom-free survival than p53 negative tumors. No difference in survival, based on p53 positivity, was observed in the glioblastoma multiforme group2.7,50 p53 in other brain tumors In contrast to astrocytomas, p53 mutations do not seem to play a significant role in the evolution of ependymoma,51 pineocytoma/ pineoblastoma52 or medulloblastoma (in which p53 mutations are found in only 5-10% of cases).53 The role of p53 in pilocytic astrocytoma25,54 is controversial. Earlier studies have reported a low mutation frequency in these tumors, whereas some recent studies that have used more inclusive mutation detection systems have reported that p53 mutations may occur in up to 35% of pilocytic astrocytoma.55 Other neoplasms in which p53 mutations do occur include gliosarcoma,56 oligodendroglioma and mixed oligoastrocytoma.57-59 Two studies on oligodendrogliomas suggest that the demonstration of p53 in tumor cells is associated with reduced patient survival.58,59 p53 in Gene therapy It was not long before the scientific community realized that p53 was ideal for therapeutic use. It was observed that radio- and chemo-sensitive tumors such as teratocarcinomas and childhood acute lymphoblastic leukemias shared the property of retention of the normal p53 gene and protein.60 In contrast, radio- and chemo-resistant tumors such as non-small cell lung cancers and colon cancers, frequently carried p53 mutations. These observations gave rise to the theory that chemotherapeutic agents killed cells by inducing apoptosis rather than by causing cellular damage61. p53 is now being used in therapeutic trials in which wild-type p53 is being introduced into tumors with p53 mutations. This maneuver is expected to restore the chemosensitivity of these tumors.62,63 Wild-type p53 has also been used as a stand-alone therapy, with the expectation that it would induce tumor cell death by senescence and apoptosis.64 So far, the vector of choice for the transfer of p53 into cells seems to be the adenovirus.64,65 Many such studies have been conducted using animal models or experimental glioma cell lines, and have shown encouraging responses to the introduction of wild-type p53, including apoptosis, inhibition of tumor growth and even tumor regression.64-66 Phase I clinical trials are currently being conducted to clarify whether specific and non-specific cytotoxic effects may occur after p53 therapy. An extended family: the "Big Brother" of p53 For almost 20 years after its discovery, the p53 gene was the only known gene of its kind, both structurally and functionally. That changed in 1997 with the discovery of the p73 gene, dubbed the "Big Brother" of p5367. Located on 1p36, this gene encodes a protein similar to the p53 protein, its talents including cell cycle arrest and apoptosis.68 Deletions of the gene locus for p73 are common in a variety of tumors, including neuroblastoma and colon and breast cancers. Much interest is now focused on this "relative" of the p53 gene. References
Copyright 2002 - the Indian Society of Human Genetics The following images related to this document are available:Photo images[hg02011f1.jpg] |
| |||||||||

{kind=link}