 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Human Genetics, Vol. 9, No. 2, July-Dec, 2003, pp. 40-50 Connexin 26 and autosomal recessive non-syndromic hearing loss Monisha Mukherjee, S. R. Phadke, B. Mittal Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical
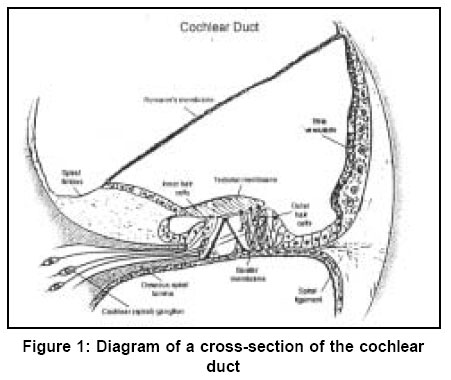
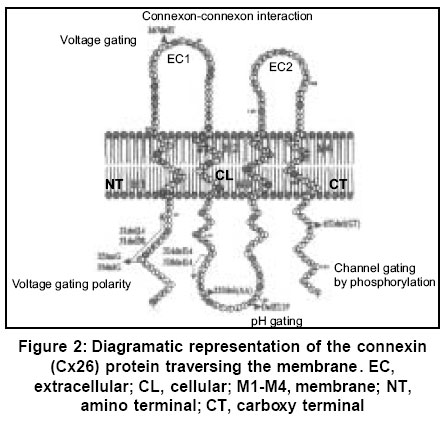
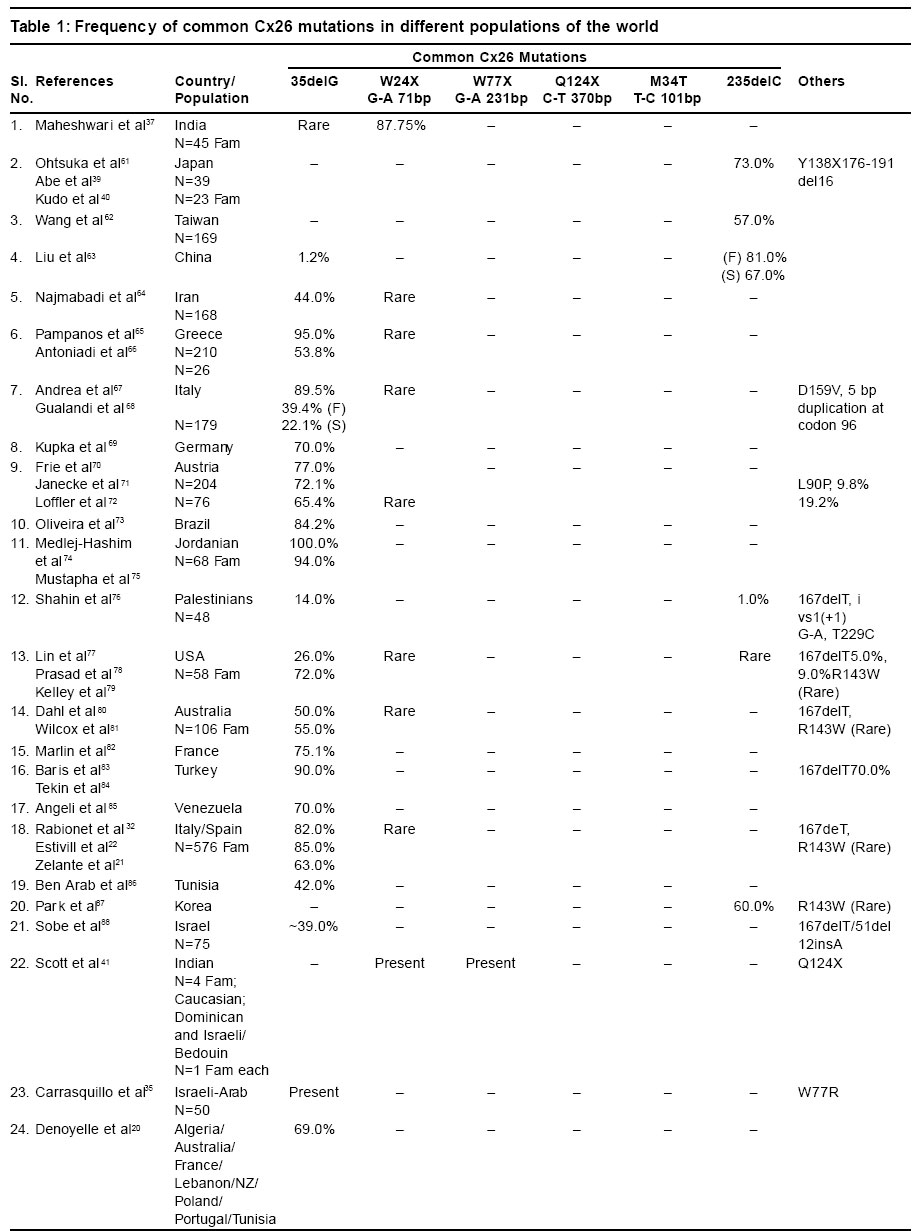
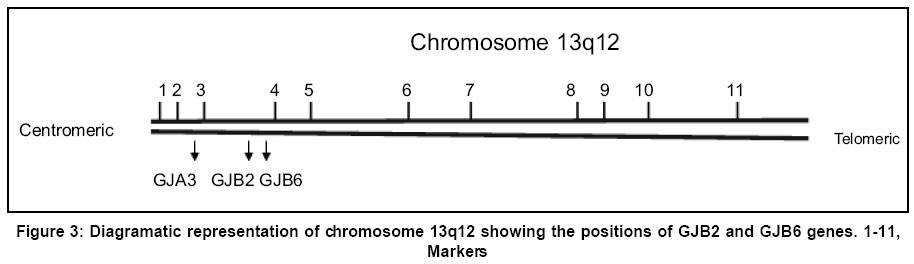
Sciences, Lucknow - 226014, India. Code Number: hg03010 Prelingual deafness occurs with a frequency of 1 in 1000 live births and is divided into syndromic and non-syndromic forms contributing 40 and 60% respectively. Autosomal recessive non-syndromic hearing loss (ARNSHL) is responsible for 80% cases of childhood deafness. Nearly all genes localized for ARNSHL cause prelingual, severe to profound, sensorineural hearing impairment. ARNSHL is genetically heterogeneous and at least 39 loci have been identified. The most significant finding to date has been the discovery of mutations in GJB2 gene at the DFNB1 locus on chromosome 13q12 as the major cause of profound prelingual deafness. This was first reported in a Tunisian family in 1994 and thereafter in many different countries. GJB2 gene encodes the gap-junction protein, connexin 26 (Cx26), mutations in which have become the first genetic marker of inherited hearing loss. Allele-specific polymerase chain reaction (AS-PCR), single stranded conformation polymorphism (SSCP) and sequencing methods have been developed for the detection of mutations in Cx26 gene. In India as well, the Cx26 mutations are being screened in families with hearing impaired children using these molecular methods. Therefore, in order to create awareness among the clinicians and the affected families; we have attempted to review the Cx26 gene mutations responsible for autosomal recessive type of non-syndromic hearing loss. The efficacy and utility of Cx26 gene analysis might open the path to proper counseling of families for carrier detection and prenatal diagnosis. It may even facilitate the development of strategies in future for the treatment of this common genetic disorder. Key Words: Non-syndromic, Hearing loss, Autosomal recessive, Connexin 26, DFNB1, mutation detection, Gap junction proteins, GJB2. Introduction It has long been recognized that heredity plays a major role in hearing impairment. Although the facts about the genetic basis of hearing loss have fascinated both clinicians and geneticists for a long time, it is only within the last few years that the genes and molecular mechanisms underlying deafness have begun to be discovered. There is a great deal of genetic heterogeneity in deafness. This reflects many different loci for deafness involved in orchestrating the hearing process.1 Hearing loss can be conductive, sensorineural or mixed in nature. Genetic heterogeneity makes the identification of causative mutation necessary for accurate diagnosis of the cause of deafness. Hearing loss is also caused by environmental factors adding complexities to identification of etiology. Genetic form of hearing loss can be syndromic or non-syndromic. The syndromic disorders are those where hearing loss is accompanied by involvement of one or several other organ systems and non-syndromic disorders, where the inner ear appears to be the only affected organ. Hundreds of syndromic forms of deafness have been described2-4 and the underlying genetic defects have been identified for many of the more common forms. Thirty percent of genetic cases are estimated to be part of a heritable syndrome. Thus, the vast majority of genetic deafness is isolated or non-syndromic. This review focuses primarily on non-syndromic autosomal recessive type of hearing loss (ARNSHL). Structure of ear Majority of the cases (~70%) of hereditary deafness are non-syndromic,5 where the inner ear is the only system affected.3,6 The inner ear regulates 2 sensory systems: the auditory system for hearing and the vestibular system for spatial orientation and equilibrium (Figure 1). The potassium ions diffuse to the stria vascularis through gap junctions formed by connexins and are secreted back into the endolymph through potassium channels thereby maintaining the mechanoelectrical transduction system. The gap junctions are clusters of intercellular channels that allow direct communication between cells.7 Six connexins form hexameric hemichannels (termed "connexons") in the endoplasmic reticulum, which are then translocated into the plasma membrane. The connexon then docks with the connexon of an adjacent cell to form a functional channel called a "gap junction". These gap junction channels mediate the diffusion of ions, metabolites and second messenger molecules between adjacent cells.8 The major proteins of gap junctions are the connexins9 that are encoded by a large gene family. The multigenic connexin family consists of at least 13 members10 sharing a common sequence of structural motifs comprising four transmembrane spanning domains, two extracellular domains, a cytoplasmic loop, and cytoplasmic amino and carboxy termini (Figure 2). There are 11 different types of connexins each of which has tissue specificity and distinct physiological properties related to the gap junction channels. Connexin 26 is highly expressed in human cochlear cells which formed the basis for looking at its involvement in ARNSHL at the DFNB1 locus.11 Non-syndromic hearing loss (NSHL) About 1 child in 1000 is born with hearing loss which is detected at infancy or early childhood5 and about 50% of cases is attributed to genetic defects.12 Non-syndromic hearing loss is further categorized by the mode of inheritance. The inheritance pattern of monogenic prelingual hearing loss is autosomal recessive in approximately 77% of patients, autosomal dominant in ~22%, X-linked in ~1% and mitochondrial in <1%.13 The majority of congenital and prelingual deafness cases with genetic causes are autosomal recessive. Typically, in non-syndromic hearing loss deafness is the only detectable symptom: however, in some cases malfunction of the vestibular system also occurs. An interesting question arises, why gene mutations lead to isolated deafness while other gene defects are associated with defects of various other systems or organs in addition to deafness. It is becoming apparent from the cloning of deafness genes that mutations in one gene can result in two or three different types of hearing loss (i.e. syndromic and non-syndromic, autosomal dominant and recessive). For example, mutation in GJB2 gene causes autosomal dominant (DFNA3) as well as autosomal recessive (DFNB1) hearing loss.11,14,15 MYO7A gene mutation causes autosomal dominant (DFNA11) and autosomal recessive (DFNB2) hearing loss.16,17 The number of deafness-causing genes is estimated to range from 50 to 100.5,18 One gene that has turned out to be a major contributor in a large percentage of non-syndromic deafness, as well as in sporadic cases, is GJB2 encoding connexin 26, a gap junction protein. In some populations, mutations in GJB2 are estimated to be responsible for 50% of severe to profound recessive non-syndromic hearing loss and for 20% of childhood genetic deafness.19-22 The carrier rate for GJB2 mutations varies in different populations, is estimated to be as much as 3-4% in some ethnic groups.19,22-24 Mapping of genes for NSHL The different gene loci for numerous forms of deafness have been called DFN (for deafness) and are numbered in chronological order of discovery. Autosomal dominant disorders have been designated as DFNA, autosomal recessive loci as DFNB and X-linked as DFN. Recessive non-syndromic hearing loss (RNSHL) accounts for approximately 40% of all childhood hearing loss.25 Thirty-nine RNSHL loci have been reported.1 The first ARNSHL locus, DFNB1 (MIM 220290) was identified by Guilford et al in 1994 which accounts for at least 50% of this type of hearing loss.4,26 DFNB1 is the most common single cause of hearing loss in European populations. Guilford et al15 mapped DFNB1 to chromosomal region 13q12-13 in 2 consanguineous ARNSHL families from Tunisia. This hearing loss was described as bilateral and profound. Their report was followed by the identification of other consanguineous families of differing ethnic origin whose deafness was linked to the DFNB1 locus.27,28 Maw et al26 tested 18 New Zealand families and one Australian family with nonsyndromic hearing loss for linkage to chromosome 13q12. The families tested showed recessive inheritance with at least 2 affected individuals born to the normal hearing parents. Nine of 18 families showed cosegregation of haplotype markers with hearing loss compatible with linkage to 13q12. In a very large consanguineous Bedouin family with profound bilateral RNSHL, the loss was linked to the 13q12 locus,27 but in a study of 27 consanguineous Pakistani families, only one case was found to be linked to DFNB1.28 Gasparini et al29 analyzed 48 Mediterranean families (30 Italian and 18 Spanish) and found an absence of linkage to DFNB1 in 21% of Mediterranean families. These studies suggest that DFNB1 might not account for the major proportion of RNSHL in at least some non-European populations. Connexin 26 Gene Mutations Mutations in GJB2, encoding the gap junction protein connexin 2611 on chromosome 13q11, have been strongly implicated in the pathogenesis of autosomal recessive non-syndromic hearing loss at the DFNB1 locus.11,21 Subsequent analysis of GJB2 has led to identification of over 70 different recessive mutations.30 There are some other autosomal recessive loci for deafness for which the genes have yet to be determined.30 Connexin 26 (Cx26) is one member of a family of related gap-junction channel forming proteins. The gap junction protein has an important role in intercellular communication by recycling of K+ ions back to endolymph following depolarization of hair cells.31 GJB2 mutations alter this function leading to deafness. Human connexins are classified either by molecular mass (26-59 kDa) or by sequence similarities into 3 groups: gap junction α (GJA), gap junction β (GJB) or gap junction γ (GJC). Sensorineural hearing loss is caused by mutations in the gap junction genes encoding the β connexins.30,32 In the human genome, the majority of β connexin genes map to 2 gene clusters at either 1p34-p35 or 13q11-q12. The genes for 20 different connexin proteins are present in the human genome.33 The first mutation proposed to be a dominant deafness allele, M34T11 has now been shown to be a recessive deafness-associated GJB2 allele. Among the recessive mutations, the single base deletion mutation, 35delG is responsible for 80% of DFNB1 related hearing loss in European and American populations. Zelante et al21 first reported this common connexin 26 mutation in Mediterranean European population. This is a mutation deleting a guanine `G' in a stretch of six guanines in the coding region of the connexin 26 gene, GJB2 at position 30-35 (35delG). This leads to a frameshift, resulting in a stop codon at position 13. The mutation results in the synthesis of a truncated 12-amino-acid polypeptide instead of the normal 226-amino-acid polypeptide. The frequency of the 35delG mutant allele varies in different populations, may be as prevalent as 4% in particular ethnic groups34 and 2.8% in the American population.19 This mutation was also reported in Israeli-Arab,35 American19 and Ashkenazi Jewish populations,24 indicating that GJB2 mutations are remarkably common as a cause of upto 50% of prelingual (early childhood) deafness. Thus, 35delG is the cause of one of the most common human genetic disorders with an incidence similar to that of cystic fibrosis.36 However, this particular mutation is not so common in the Indian population.37 Reports are available that out of 82 recessive non-syndromic hearing loss (RNSHL) families (51 Italian and 31 Spanish), up to 50% had mutations in Cx26.22 Isolated cases of Cx26-associated hearing loss have been reported from northern and central Africa as well.38 Among 58 American families diagnosed with RNSHL, 20 families were found to have mutations in both alleles of Cx26.19 The two most common alleles were 35delG (thirty-three) and 167delT (nine). In a study of an Ashkenazi Jewish population with RNSHL, both 35delG and 167delT were found.24 The prevalence of the 167delT mutant allele in the normal Ashkenazi Jews was found to be 4.03%, while that of 35delG was 0.73%. A survey of 32 Japanese RNSHL families found no 35delG mutation.39 However, a different frequent new mutation, 235delC was detected.39,40 There are three mutations in the GJB2 gene in Japanese patients with childhood deafness: 2 frameshift mutations, 235delC and 176-191del,40 and one nonsense mutation, Y136X. Each of the three mutations results in truncated protein and may disrupt the recycling of endolymphatic potassium ions. The frequent presence of 235delC in Japanese patients suggests that for this mutation there may also be a founder effect. The frequency of some of the common Cx26 mutations present in different world populations has been described in Table 1.30 Mutation 35delG has a carrier frequency in the Caucasoid population of 1/31 and 1/3.22,34 Mutation 167delT has a carrier frequency in the Ashkenazi Jew population of 4% while frequency of 35delG in this population is 0.7%.24 The frequency of mutation 235delC in the Japanese population has been determined as 2/203.45 The carrier frequency of mutation M34T in the Belgian population is 2.4%. Another two carrier screenings for M34T on Caucasoid populations reported a frequency of 3/192 and 1/200 respectively.19,23,41 Although it has been proposed that a succession of six Gs may present a mutational hotspot,19,20,24,35 a recent study has provided evidence that the high frequency of this mutation is the result of a founder effect.42 Other defects identified are scattered through out the gene. They include nonsense mutations (n=6) and small deletions/insertions (n=8), most of which lead to a premature termination of protein translation. In addition, several missense mutations (n=6) have been reported in compound heterozygotes, these mutations have been assumed to be pathogenic, although no studies have demonstrated defective connexin function. These data provide strong evidence that loss of functional Cx26 underlies non-syndromic recessive deafness and have established GJB2 as the DFNB1 gene. Other Complexities of Connexin 26 The phenotype of the Cx26 induced hearing impairment is variable because the degree of hearing loss varies from mild to profound. Moreover, it exhibits intrafamilial and interfamilial variability. Different genotypes of Cx26 mutations have been found in the families of multiple affected members. There are many reports finding no correlation between genotype and phenotype.43,44 It remains unclear why mutations in Cx26 gene lead to different degrees of hearing loss. It has been hypothesized that additional mutations in other interacting genes can influence the extent of hearing impairment.45 The presence of heterozygotes for connexin 26 in deaf people has increased the importance of relevant molecular understanding and diagnosis of hearing loss and appropriate genetic counseling. Heterozygosity for a mutation at the GJB2 locus in patients with prolonged hearing loss has been observed. Various explanations proposed for this include the possibility of a mutation in the non-coding region of the gene or involvement of another gene.46,47 Other Connexin Genes Auditory function is an extraordinarily complex process. To date, expression of at least four different connexins other than Cx26 has been reported in the inner ear i.e. Cx32 (GJB1), Cx30 (GJB6), Cx31 (GJB3) and Cx43 (GJA1). The GJB1 (Cx32) is also responsible for X-linked Charcot-Marie-Tooth disease type I; GJB3 (Cx31) is involved in deafness or a skin disease, erythrokeratoderma variabilis, depending on the location of the mutation; GJB6 (Cx30), has been related to a dominant type of deafness in an Italian family and GJA1 (Cx43), has recently been shown to be involved in recessive deafness. GJB2 mutations account for a significant proportion of ARNSHL and the number of remaining deafness loci is large. This makes it unlikely that another gene will be a common cause of recessive deafness. Most of the mutations in GJB2 gene lead to complete absence of connexin 26. Though GJB2 is expressed in a diversity of tissues, its mutations are not associated with defects of any other system other than deafness. It means that other connexins can substitute for Cx26 in these tissues but not in the cochlea.48 These findings suggest that the expression of Cx26 in the cochlea is essential for audition and that other connexins cannot compensate for the loss of Cx26 in the auditory epithelial cells. Cx26 and Cx30 are constituents of both epithelial and connective tissue gap junction systems and appear to have very similar patterns of distribution and function.31,49 Large deletions in the GJB6 were detected which cause recessive non-syndromic deafness in the Mediterranean or Ashkenazi Jewish populations.47,50 It was suggested that this recessive mutation caused hearing loss in individuals that are double heterozygous for the deletion and a mutation in the GJB2 gene, either due to a digenic mode of inheritance or because the deletion abolishes control elements that are important in the expression of GJB2. Since Cx30 and Cx26 are close to each other on chromosome 13q12 (Figure 3), a 342 kb deletion i.e. loss of Cx30 allele was proposed to have a cis silencing effect on the adjacent Cx26 allele.47,48 This deletion has been reported to cause deafness both in homozygous status and in heterozygosity with a GJB2 point mutation in trans. Mouse model studies suggest that the absence of Cx30 is sufficient for the severe deafness in some patients who have been found to carry only the large deletion in a homozygous state (i.e. with two intact Cx26 alleles).47 Cx43 has also been found in both gap junction systems.51 Cx31 expression in the mature auditory organ is still a matter of debate.52 The functional importance of these gap junction networks is that all four mutations can cause hearing loss.11,20,47,50 However, their roles in the cochlea are poorly understood. Analysis of Cx26 mutations GJB2 screening has become widely available, in part because the small size of the single coding exon facilitates gene sequencing. PCR-based sequence analysis has been shown to be an efficient method for identifying pathogenic mutations in this gene and is rapidly emerging as the standard of care for the evaluation of newborn infants as well as older children and adults with non-syndromic deafness of uncertain causes.53 Molecular Testing for Cx 26 Mutations The genetic diagnosis of GJB2-related deafness is dependent on identifying mutations within the DNA of affected individuals. DNA is extracted from peripheral blood samples using standard protocols. Mutation screening can be completed using a variety of techniques. Samples from all probands can be screened for GJB2 (Cx 26) mutations by using PCR amplification of genomic DNA by allele-specific primers.41 The PCR products are analyzed by electrophoresis on a 1.5% agarose gel containing ethidium bromide. Heterozygotes at the GJB2 locus should be screened for the GJB6 deletion as a cause of deafness. The GJB6 deletion can be screened by using the method and primers described by del Castillo et al.47 Among the huge number of connexin mutations there are a few (5-6) most common ones which are screened by allele-specific polymerase chain reaction (AS-PCR) viz. 35delG (del of G at 30-35), W24X (G to A at 71), W77X (G to A at 231), Q124X (C to A at 370), M34T (T to C at 101).41 Primers for these particular mutations can be used for the initial screening for ARNSHL in any population. The common mutations are identified through AS-PCR and the presence of putative mutations is confirmed by sequencing. In case the common mutations are not detected by AS-PCR, the amplification products are analyzed in single-stranded conformation polymorphism (SSCP) assays followed by sequencing. PCR is performed with genomic DNA using the primers according to Scott et al.41 PCR products are electrophoresed on a 6% SSCP gel (49:1) for approximately 8 hrs at 20 W. The resulting gels are dried and visualized by autoradiography. Due to the relatively small size of the Cx26 coding region and its location on a single exon, PCR amplification and SSCP are followed by direct sequencing and can be used as initial screen for mutations. The products obtained after PCR amplification using primers for exon 1 and exon 241,54 are electrophoresed in 2.5% low melting agarose gels, excised and purified using QIAEX II Gel extraction Kit (Qiagen, Hilden, Germany). The products are subjected to ABI PRISM Big Dye Terminator cycle sequencing kit, the sequenced products are purified and loaded onto ABI Prism 310 Genetic Analyzer (PE Applied Biosystems, Foster City, CA). The sequences are analyzed using suitable software programmes.55,56 Diagnosis and Screening Prevalence studies of Cx26 mutations in persons with severe to profound or profound ARNSHL have shown two unexpected findings. First, in a large number of ethnically different populations throughout the world, Cx26 mutations are responsible for more than half of the cases of ARNSHL.19,22,36 Second, in many of these populations a single mutation, the 35delG, predominates.19,22,36,41 These data have developed an interest amongst the clinicians since the high frequency of the 35delG mutation will facilitate molecular diagnosis. The identification of the 35delG mutation early after birth will enable families to modify the educational process of the deaf child. Since the frequency of this particular mutation varies from one population to the other, new mandates for hearing, screening programs for newborns and new information on the genetics of hearing loss can be used to diagnose the cause of hearing loss in children and also understand better the molecular biology of hearing. The molecular diagnostic test should allow the determination of a genetic origin in about a third of the unexplained prelingual sporadic cases of deafness. Individuals with congenital deafness should be offered Cx26/GJB2 screening to establish an etiology and to provide prognostic, genetic and therapeutic information. Effective rehabilitation for profoundly deaf individuals with GJB2 related deafness is possible through cochlear implantation.56,57 Genetic Counseling The discovery of mutations in GJB2 has significant implications in the early diagnosis, management and genetic counseling for recessive and sporadic congenital deafness. The genetic counselors will have the necessary information to provide better recurrence-risk data to parents of a hearing-impaired child. Carrier detection in relatives of affected and carrier parents would be feasible, leading to prevention of the disorder and early treatment of affected people. Since only 60% of deaf individuals are homozygous, a more extensive analysis of Cx26 will be necessary in a high proportion of cases to distinguish the common heterozygous carriers for Cx26 mutations from DFNB1 affected children. Very recently Pandya et al54 reported that molecular testing for GJB2 and GJB6 should be offered to all patients with non-syndromic hearing loss since GJB2 mutations have been shown to cause deafness when present with a deletion of the GJB6 gene. The variability of hearing loss associated with Cx26 gene mutations is considerable even within families.58 It is likely that environmental influences and modifier genes are playing a role in the variability of hearing loss. This makes genetic counseling more difficult. Linkage of Cx26 testing to newborn screening programs will be important in identifying this major cause of deafness. Cx26 analysis should be a routine test recommended to families with infants/children identified as having non-syndromic deafness of unknown etiology in the neonatal period and beyond. The continual discovery of genes mutated in hearing loss has led to the need for adequate and careful evaluation for diagnosis and possible treatments both by prenatal and postnatal genetic testing. This opportunity has given rise to a host of ethical issues for individuals with hearing loss who have become a part of the `deaf culture'. They think that their culture will be threatened by genetic studies. An appropriate interpretation and counseling for any genetic testing are important, and of course personal decisions ultimately rest with the individuals and families for whom these services are being provided. Advantages of GJB2 Mutation Screening Testing for DFNB1 mutations as the first step in determining the cause of hearing loss has many advantages. A positive result for biallelic mutations in the GJB2/DFNB1 gene should eliminate the need for expensive tests such as electroretinography and tests using radiation thereby significantly reducing medical costs. There will no longer be the need for deep sedation or general anesthesia of children for many of these tests. Therefore, coupling Cx26 testing for newborns with hearing loss established by diagnostic Auditory Brainstem Response Audiometry will have significant utility for the parents and professionals caring for these children. Knowledge of genotype will allow physicians, audiologists, educators and geneticists to counsel parents more appropriately. Genetic counseling of parents with sporadic patients with Cx26 hearing loss allows accurate statement of risk that a subsequent child might have similar hearing loss. In states where there are universal newborn screening programs and difficulty arises after failed diagnostic auditory brain-stem response testing, confirmation of the Cx26 mutation may help prioritize follow-up. The parents who want to know the source and recurrence risk of their child's hearing loss will thereby receive an answer. Availability of molecular diagnostic facility for Cx26 screening has made prenatal testing feasible. Conclusions Genetic testing may offer valuable information regarding decisions for educational approaches, choices in modes of language acquisition, including sign or spoken language, career and lifestyle decisions, and potential intervention and treatments such as amplification devices or cochlear implants. Matsushiro et al59 reported the successful cochlear implantation (CI) in patients with prelingual deafness and 235delC mutation. The molecular genetic testing promises improved diagnosis of sensorineural hearing loss in children; it also helps in resolving the diagnostic dilemmas due to variable phenotypes. However, it is important to consider the ethnic background when performing genetic testing because different mutations exist in different ethnic groups. Apart from the immediate implications of genetic studies, other scientific endeavours in the molecular genetics of the sensory perception of the inner ear are crucial for solving the mysteries of complex biological processes. Detailed molecular studies will undoubtedly lead to a more fundamental and thorough understanding of the development, structure, and function of the auditory and vestibular systems that serve as critical gateways to an enriched perception of a complex set of stimuli in our outside world. The pace at which additional genes are discovered is expected to increase in the coming years because of the availability of cochlea-specific cDNA libraries and completion of the sequencing of the human and mouse genomes.60 Knowledge of a gene commonly involved in congenital deafness will lead to the development of animal models, which should be useful for the study of the pathophysiology and development of strategies for therapeutic intervention. Acknowledgements One of us (MM) is thankful to CSIR, New Delhi for providing senior research fellowship. References
Copyright 2003 - the Indian Society of Human Genetics The following images related to this document are available:Photo images[hg03010t1.jpg] [hg03010f2.jpg] [hg03010f1.jpg] [hg03010f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}