 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
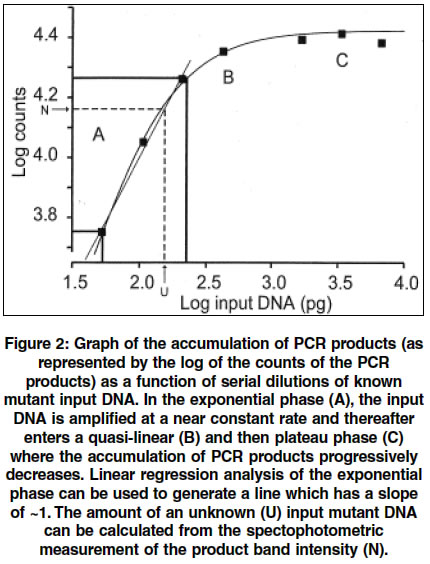
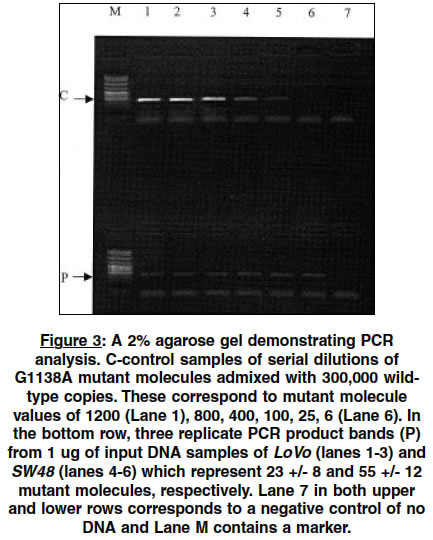
Indian Journal of Human Genetics, Vol. 10, No. 1, Jan-June, 2004, pp. 13-17 Original Article A simple and rapid quantitative method of detection of the common achondroplasia mutation: Analysis in mismatch repair deficient cells Grewal Raji P New Jersey Neuroscience Institute, Edison, New Jersey - 08816 Code Number: hg04004 ABSTRACT Achondroplasia is the most common form of dwarfism and has an incidence of approximately 1/7,500. In more than 97% of cases, it is caused by a recurrent point mutation, a G to A substitution at nucleotide position 1138 (G1138A) of the fibroblast growth factor receptor 3 gene. Although this is an autosomal dominant condition, more than 90% of all mutations occur sporadically making this one of the most mutagenic sites in the human genome. The reasons for the high spontaneous G1138A mutation rate are not known. This investigation was performed by developing a simple and rapid semi-quantitative allele specific PCR based assay capable of reliably detecting more than 25 mutant G1138A copies in a pool of 300,000 wild type molecules. Using this assay, the G1138A mutation frequency was measured in cell lines deficient in mismatch repair (LoVo, SW48) and comparing it with controls. No differences were found in the frequency of this point mutation between the mismatch repair deficient and wild type cell lines.INTRODUCTION Achondroplasia (ACH), the most common form of dwarfism is an autosomal dominant disorder with an incidence of approximately 1/7500.[1],[2] In more than 97% of patients with ACH, the disease is caused by a single, recurrent point mutation resulting from a G to A substitution at nucleotide position 1138 (G1138A) in exon 10 of the fibroblast growth factor receptor 3 gene (FGFR 3) (3). Moreover, in more than 90% of cases the condition is sporadic and represents a spontaneous germline mutation.[2],[3] The reported disease prevalence ranges from of 1/15,000 to 1/77,000 and since more than 90% of these cases represent new mutations, the mutation rate at the FGFR3 1138 guanosine nucleotide is between 5.5 x 10-6 and 3.2 x 10-5 per gamete per generation.[3] On average, at other similar nucleotide sites in the genome, mutation rates for CpG-to-TpG transitions have been estimated to occur at a rate of 3.68 X 10-8 to 1.05 X 107 per gamete per generation. This places the estimates of the mutation rates at this site to be between 50 and 760 times higher than previously estimated and one of the most mutable sites in the entire human genome.[3] The reasons for the apparent high rate of mutation at this site are unknown. Point mutation rates have been shown to differ significantly among regions of the mammalian genome. The factors which determine these differences are complex and could include local sequence variations, the presence of pseudogenes, C/G content, undefined features of local chromatin content and differential germline methylation of cytosines in CpG sites.[4],[5],[6],[7] More recent studies have explored the relationship between DNA repair and mutation frequencies.[8],[9],[10],[11] In general, the study of point mutations has been limited by the availability of reliable assays that are capable of detecting mutant molecules in a pool of wild type molecules. By developing a rapid and sensitive assay, the effect of loss of some forms of mismatch repair (MMR) on the mutation rate at the G1138A site was studied. MATERIALS AND METHODS The mutation was measured by a semi-quantitative assay developed using an allele-specific polymerase chain reaction (ASPCR). ASPCR is based upon the principle that alleles differing by one or more nucleotides can be distinguished on the basis of PCR amplification.[12] The general technique involves generating primers that have mismatches at the 3′-end of the primer. However, the system is not absolute and in some cases, the mismatches may amplify.[13] It has been demonstrated that introduction of additional mismatches can alter the ASPCR primer/wild type template DNA complex and render them increasingly refractory to extension[14] [Figure - 1]. To obtain maximal specificity and sensitivity, PCR conditions such as primer, nucleotide and enzyme concentrations and cycling parameters are modified. In PCR, each cycle consists of denaturing the target DNA, annealing of primers and their extension along the template DNA via a thermostable DNA polymerase. The newly synthesized DNA segment can serve as a template in subsequent cycles resulting in exponential amplification of the original target DNA. There are three distinguishable phases of PCR. During the exponential phase, the target DNA is amplified at a nearly constant exponential rate[15] [Figure - 2]. Thereafter, in the later cycles, the synthesis rate drops dramatically and enters a quasi-linear phase. Eventually, a plateau phase is reached which is defined as the attenuation in the rate of the exponential product accumulation. The accumulation of PCR products in the exponential phase of the reaction can be modeled by the equation, N = n(1+E)k where N is the number of molecules at the end of the reaction, n the number of template molecules, k is the number of cycles, and E is the average efficiency. Therefore, there is a linear relationship between the logarithm of the amount of initial template and the final amount of the specific, amplified product: log N = log n + k log(1 +E), as reviewed by Soong and Arnheim.[15] For this to be true, E must have a value of close to 1, which occurs only during the exponential phase, where the number of PCR products doubles during each cycle. The linear relationship can be used to determine the quantity of an unknown amount of starting template. This can be done by simultaneously amplifying known amounts of starting template and using the data from the amplification signals of these known samples to create standard curves. The amount of target DNA in different unknown samples can then be quantitated by comparing the amplification signals of the samples to the standard curves. After a systematic survey of annealing temperatures and times, nucleotide concentrations, different polymerase enzymes, thermocylcers and a series of different primers, a highly specific and sensitive assay for the G1138A mutation was developed employing ASPCR. First, 10 rounds of non-specific PCR were carried out using primers [Figure - 1] to proportionately increase the copy number of both the ACH mutation and wild type templates. This was performed under the following conditions: 0.2 mM dNTP, 10% (V:V) DMSO, 50mM KCl, 10mM Tris-HCl, 2.5 mM MgCl2 and 2.5 units Taq DNA polymerase (Promega). The primers for this round of PCR have been previously described[1] and used at a concentration of 0.5uM GR1-5′-GGAGATCTTGTGCACGGTGG-3′ and GR2-5′-AGGAGCTGGTGGAGGCTGA-3′ [Figure - 1]. The reaction was run in a final volume of 50 ul, overlaid with 50 ul of mineral oil and subjected to PCR under the following cycling conditions (thermocycler-MJ Research):95°C- 2min; 94°C,-45 sec, 65°C-45 sec, 72°C-30 sec, X 9 cycles then 72°C-3 minutes and 4°C. A small aliquot was then taken and amplified with the G1138A mutation allele specific primers that were designed [Figure - 1]. Five ul of this reaction product was diluted in 195 ul of water and 2 ul of the dilution was then subjected to a second round of allele specific PCR under the following conditions: 0.05 uM of P1-5′-ATGCAGGCATCCTCAGCTCCA-3′ and P2-5′GGGAGATCTTGTGCACGGTG-3′, 0.08 mM dNTP, 1X Amplitaq Gold buffer, 1 unit of Amplitaq Gold (Applied Biosystems). The cycling parameters were as follows (thermocycler-Perkin Elmer 9600): 95°C-10 minutes; 95°C-40 sec, 66.5°C-10 sec, 72°C-15 sec for 35 cycles followed by 72°C -3 minutes and then 4°C. Ten ul of the reaction product was then electrophoresed in a 3% agarose gel at 100 mV for about 30 minutes, stained with ethidium bromide and an image obtained using an imaging system (Eagle Eye II still video system, Stratagene). To test the specificity and sensitivity of the PCR reaction, ACH patient DNA was mixed with a clone containing a of fragment wild type ACH. Both of these ACH sequences were confirmed by sequencing, and quantitated by spectrophotometric analysis. Cloned wild type ACH was used as a control because all somatic DNA samples gave the ACH mutation specific PCR product. A series of experiments were then performed with serially decreasing numbers of mutant ACH copies admixed with the 300,000 wild type clone (equivalent to 1 ug of human genomic DNA) copies. The results indicated that with amplification of an admixture of a minimum of 25 mutant ACH genome equivalents mixed with 300,000 wild type ACH copies, the expected 128 bp product was observed. Below 25 ACH copies, this product was not consistently observed indicating the minimum sensitivity of this assay. For each PCR reaction, control standards consisting of 400, 100 and 25 genome equivalents of ACH patient DNA mixed with 300,000 copies of the cloned wild type ACH fragment were assayed with a negative control (no DNA). Simultaneously, 2 samples of 1 ug of DNA where the mutant copy levels were unknown were assayed in triplicate and the experiment was performed using the same reagents for all samples. The images of the electrophoresed products from all experiments were visually inspected to determine suitability for further analysis. Features that were analyzed included specificity of the reaction (one product band observed), absence of a product in the negative controls and similar band intensities among the three replicates of each unknown. The acceptable images were stored and the intensity of the product bands were quantified (EagleSight Software, Stratagene). A plot of the logarithm of the intensity of the mutation-specific band as a function of the amount of the logarithm of the ACH mutation-containing input DNA was generated. If the slope of the linear phase of the generated curve was approximately one, the data was suitable for quantitation. The cell lines (LoVo, SW48, SW837, HT-29) used in these experiment were obtained from American Type Culture Collection and grown to confluence as recommended. They were harvested by standard methods and DNA was extracted as previously described and all samples were quantitated using the same spectrophotometer. Only those samples where the 260/280 ratio was greater than 1.7 were assayed. RESULTS As indicated in [Figure - 3], the analysis shows the results of a reaction in the exponential phase. The calculated values indicate mutant copy levels of 23 +/- 8 (LoVo) and 55 +/- 12 (SW48). The calculated values in the control cell lines are 31 +/-10 (HT-29) and 49 +/- 15 (SW837) copies (data not shown). DISCUSSION These experiments demonstrate the utility of this ASPCR assay which is reliable, rapid and inexpensive. The validity of this assay has been independently confirmed by the development of a kinetic PCR based system (unpublished data on the frequency of the G1138A mutation in sperm DNA.[16] In contrast to the kinetic PCR, ASPCR requires minimal equipment that is already present in a molecular genetics laboratory and overall is relatively inexpensive to perform. It may be useful as a screening assay in model systems designed to investigate the unusually high mutation frequency at the G1138A site. The results indicate that there is no difference in the frequency of the G1138A mutation in the cell lines deficient in MMR compared to controls. The two cell lines studied, LoVo and SW48, have deficiencies of hMSH2 and hMLH1 of MMR, Respectively.[15] These proteins are the human homologs of E. Coli proteins, mut S and mut L, that constitute part of the MMR complex. In E. Coli, mismatches are recognized by this complex and the incorrect base is excised in a fragment of 1000 bp of surrounding sequence. In humans, much less is known about the mechanisms of MMR. This absence of an increase of the mutation rate in these cell lines could represent a redundancy that exists in the DNA repair system.[18] However, there are alternative explanations. It may be that a selection process occurs in which those cells that harbor this mutation are less viable and die. In addition, the degree and timing of methylation may also contribute to the mechanism of mutagenesis at this CpG site. Finally, there may be other mechanisms which are unique to germline tissues that contribute to a selectively high mutation rate compared to somatic tissues. The study of G1138A mutation is important and may provide insight into fundamental mechanisms of mutation. Such insight may have implications not only for medical science but the evolution of genes. ACKNOWLEDGEMENTS I thank D. Cohn for providing the human wild type and control achondroplasia DNA and N. Arnheim for advice in developing the assay. REFERENCES
Copyright 2004 - Indian Journal of Human Genetics The following images related to this document are available:Photo images[hg04004f3.jpg] [hg04004f2.jpg] [hg04004f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}