 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
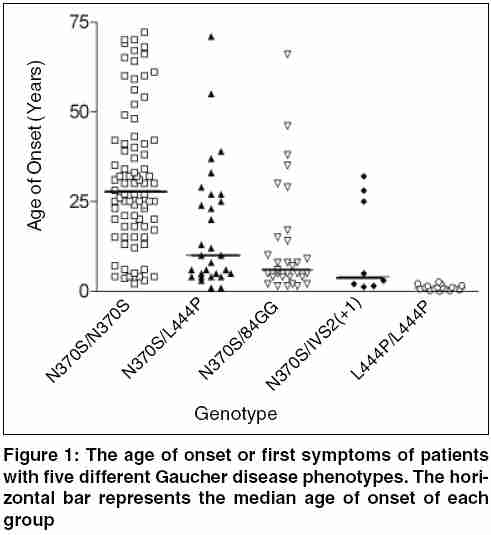
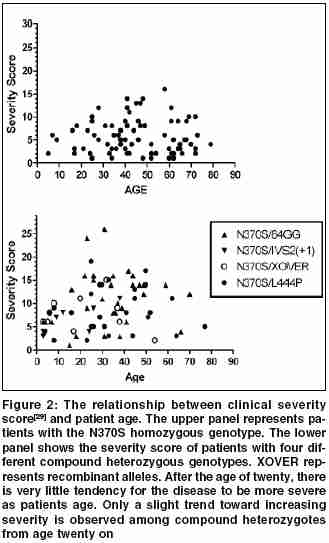
Indian Journal of Human Genetics, Vol. 11, No. 3, September-December, 2005, pp. 121-127 Invited Article The treatment of Gaucher disease in countries with limited health care resources Beutler Ernest The Scripps Research Institute, Department of Molecular and Experimental Medicine, 10550 North Torrey Pines Road, La Jolla,CA 92037, USA Code Number: hg05024 Gaucher disease is probably the most common of the lysosomal storage disorders. Mutations of the gene encoding glucocerebrosidase result in accumulation of the substrate of this lysosomal enzyme, viz. glucocerebroside. As a result this glycolipid accumulates principally in the macrophages in type 1 disease, and in the neuronopathic forms of the disease (type 2 and type 3) in the central nervous system as well. Type 1 disease is by far the most common and is characterized by hepatosplenomegaly, thrombocytopenia, and often skeletal involvement. Historical Considerations The past 15 years has seen major advances in the management of patients with this autosomal recessive disease. These include the availability of enzyme replacement therapy, and more recently what has been designated as substrate reduction therapy. Enzyme Replacement Therapy The concepts behind enzyme replacement are not new. The idea that lysosomal storage diseases could be treated by exogenous administration of enzyme was first suggested by de Duve more than 40 years ago,[1] when he wrote:"Any substance that is taken up intracellularly by an endocytic process is likely to end up within lysosomes. This obviously opens up many possibilities for interaction, including replacement therapy." Attempts to implement this strategy were carried out in the 1970s by Roscoe Brady′s group at the NIH[2] and by our group, and then at the City of Hope in Duarte, California. [3],[4],[5],[6],[7] Although we were able to document some encouraging clinical results, the method of delivery that we used, viz. encapsulating enzyme in red cell ghosts, was so cumbersome as to render this treatment approach impractical. The demonstration by Achord and Sly[8] in 1978 that macrophages have a mannose receptor that could be used to target exogenous enzyme to the cells was a major breakthrough. With the involvement of industry (Genzyme Inc.) it now became possible to make a targeted enzyme on a commercial scale and this enzyme was found to be very effective in treating type 1 Gaucher disease.[9],[10] Substrate Reduction Therapy The suggestion that inhibition of glucocerebroside formation may be helpful in treatment of this disease was first made verbally by Radin in the 1970′s and in publication form almost 20 years ago by his group[11] and others.[12] The implementation of substrate reduction therapy was the indirect outcome of an unsuccessful trial of an enzyme inhibitor in the treatment of HIV infections. This inhibitor, now known as miglustat, is directed against the enzyme that transfers glucose to ceramide forming glucosylceramide (glucocerebroside). It had generally been thought that the use of such inhibitors, blocking the action of an enzyme so essential in glycolipid synthesis, would have prohibitively toxic side effects. But the use of this material in HIV infections made it clear that its toxicity was much lower than had been anticipated.[3],[4] The initial clinical trial showed that this inhibitor had distinct clinical activity in patients with Gaucher disease, reducing the size of both liver and spleen.[15] These investigations were confirmed in subsequent studies. The drug is somewhat less effective than enzyme replacement therapy, and has more side effects. These include gastrointestinal symptoms, neuropathies and tremor, all of which are believed to be reversible. Cost of Treatment as a Problem for Countries with Limited Health Care Budgets In reality, all countries have limited annual health care budgets. This includes the United States which spends a staggering $5267 per person for health care.[16] But the budgets of countries with per capita incomes of under $1200 is only $26. The usual initial dose of Cerezyme®, the enzyme preparation used to treat Gaucher disease, is 130 units per kilogram body weight per month. For a 50 kg child obtaining therapy at this dose level the cost is approximately $300,000 per year for the medication alone. This represents the entire health-care budget for 11,000 citizens in the average country with a per capita income of under $1200. And how many patients will have to be treated in such a country? Data from Portugal,[17] Italy,[18] and Australia[19] suggest that the birth frequency in these, essentially European, populations is about one per 50,000. One can calculate, then, that at this prevalence of the disease countries with a per capita income of under $12,000 would spend one-fourth of their entire health care budget on treatment of this disease, clearly an untenable situation. Brazil, a country with a per capita health expenditure of $271 (US) in 1998[20] currently expends $49,000,000 per annum for the treatment of Gaucher disease,[21] an amount equal to the total healthcare expenditure of nearly 181,000 inhabitants to treat 425 patients. The cost was higher until efforts were made to reduce the high dosages that were given to patients.[21] The gene frequency for Gaucher disease mutations is very high in the Ashkenazi Jewish population, approximating 0.037,[22] and therefore Israel, with a population with a high proportion of Ashkenazi Jews is a special case. Although health care expenditures in Israel are estimated at $1622 per capita (http://www.euro.who.int/eprise/main/who/progs/chhisr/system/20050131_1), this relatively large expenditure is counterbalanced by the fact that it has a large Ashkenazi Jewish population, which has been estimated at 3,700,000 (http://i-cias.com/e.o/jud_ashk.htm). Based on the Hardy-Weinberg equilibrium, one could estimate that the birth incidence of patients with the Gaucher disease genotype would be 1:730-over 5,000 patients in the country. At an expenditure of $300,000 per year, treatment of each patient with a diagnosis of Gaucher disease would consume the health care budget of nearly 1,000,000 citizens and, the total cost to the nation would be a staggering $1.5 billion dollars a year. Clearly, this is an untenable situation, which has not come to pass. Indeed, the sales of Cerezyme are approximately $1 billion dollars a year (http://www.clearclicks.com/news/Business+News_124/From-orphan-to-blockbuster-_151923.html) - sales by Genzyme are estimated to top $870,000,000 this year, and this does not take into account the markups in price in the delivery chain. Even this is a major strain on the health care system of many nations. But the assumptions that we have made here is that every patient is treated with a frequent program that cost $300,000 a year, and that treatment is carried out solely with modified glucocerebrosidase sold by Genzyme Corporation. It is my purpose to review the alternatives that exist. These alternatives cannot solve the problem, but they can certainly ameliorate it. Partial Solutions to the Problem Treat Only Those Patients Who Really Need It It has sometimes been implied that all patients with Gaucher disease should be treated to "prevent" the development of complications of the disease.[23] An understanding of the natural history of this disease makes it clear that such an approach is based upon misconceptions about the natural history of the disease. Some patients with Gaucher disease, especially children, manifest rapid progression of symptoms. These are the patients who need treatment. They tend to be the ones that we, as physicians, tend to remember, but they do not represent all patients with Gaucher disease by any means. In some populations they are, in fact, a distinct minority. There have been relatively few studies of the natural history of Gaucher disease. [24],[25],[26],[27],[28],[29] These studies show that among adults with the homozygous c.1226A>G (N370S) mutation there is little or no progression in the vast majority of cases. In patients who are compound heterozygotes for a mild mutation such as N370S, and a more severe mutation such as c.1448G->C (L444P), progression is variable. As shown in [Figure - 1], it is notable that the homozygotes for the N370S mutation tend to have a much later age of onset than the compound heterozygotes or homozygotes for the L444P mutation. Moreover, when the "severity score" is plotted against the age of patients, it is notable that there is little increase in severity of disease with increasing age, particularly among the N370S homozygotes. [Figure - 2] Among Ashkenazi Jewish patients, because of the high prevalence of the N370S mutation, mild disease tends to be the rule, while in non-Jewish populations more severe disease phenotypes are encountered. Even in the latter populations, however, there are numerous patients who do not need treatment. Do Not Overdose Your Patients It has been commonplace to initiate therapy with infusions of 60 units per kilogram every two weeks, 130 units per kilogram per month. But why was this dose chosen? It was simply that when the initial clinical trials were performed[9] the investigators very reasonably used a dose that was sufficiently large to ensure success. The accumulated evidence shows, however, that satisfactory clinical results can readily be obtained with as little as 1/10 of this dose. [Figure - 3] depicts the results of a meta-analysis of all published data in which evaluation of response was carried out after six months of treatment. It is apparent that 30 units per kilogram per month gives results that are equivalent to those obtained with 130 units per kilogram per month. Even one-half of this dose seems to give adequate, although slightly suboptimal, responses if the enzyme is administered three times weekly, a schedule that seems much more efficient for the delivery of an enzyme that is designed to bind to a relatively small number of high affinity receptors on macrophages. The proposal has also been made that therapy be initiated with a very low dose, 15 units per kilogram per month and that at next month intervals the patient be evaluated and the dose doubled or reduced to one half depending upon response.[34] Reasonable as this approach may appear, it is based upon the hypothesis that the variable response of patients to therapy is a result of differing sensitivities to therapy. If this were the case, one would expect that the number of treatment "failures" with small doses would be greater than with larger doses. As shown in [Figure - 4], meta-analysis of all of the available data using the criteria for "failure" employed by Hollak et al[34] shows no evidence of dose sensitivity. It has often been suggested that while "low dose therapy" may be adequate for patients with mild disease, that treatment of severe disease requires a larger dose. Again, the facts do not bear out this contention. Using liver volume as a readily measured and highly relevant surrogate for disease severity, a meta-analysis shows that the shrinkage of even very large livers is equivalent at doses ranging from 30 U to 130 U/Kg. [Figure - 5] My recommendations, then, is that patients be treated with 15 units per kilogram per month, and that this dose should be divided so that it is given at least weekly, and preferably three times a week. The response to this dose seems to be somewhat less than that obtained with 30 units per kilogram per month, but one can then treat twice as many patients at nearly the same cost. The arithmetic is simple: for a 70 kg patient at a price of four dollars per unit of enzyme, the annual cost is $436,800 at a dose of 130 units per kilogram per month, $201,600 at a dose of 60 units per kilogram per month, and only $50,400 at the dose of 15 units per kilogram per month. This saving can make an enormous difference in the number of patients that can be treated. Finally, resources can be conserved by judicious use of "drug vacations". It is conventional wisdom that patients need to be on some type of maintenance therapy to prevent rapid progression of their disease once treatment has been successful in eliminating most of the stigmata of the disorder. But there is no evidence that this is true, or that it is true for all patients. It has been our experience and that of others[41],[42] that once the patient is in a state in which the disease burden is minimal it is often possible to discontinue therapy for months or even years. When organomegaly or hematologic changes begin to recur there is ample time to reinstitute treatment. Obviously, suspending treatment saves resources, but it is also psychologically beneficial for many patients to be told that they have responded so well that they can suspend therapy for a time. Administering a Substrate Inhibitor Although low molecular weight inhibitors of glucocerebrosidase are undoubtedly inexpensive to synthesize, miglustat is being sold at a high price. Consequently, little saving can be achieved by using these potentially less expensive treatment modalities. If the price of inhibitors were lowered substantially they might prove an excellent alternative to enzyme replacement therapy in some patients. But at present, they are as costly as enzyme replacement when the appropriate low dose therapy regimen is given. It is notable in this regard that while the dose of enzyme usually given in some Western countries is much higher than is needed, reducing the dose of miglustat has been shown to severely impair its effectiveness.[43] Making Your Own Enzyme Preparation It is conceivable that some nations, perhaps with the cooperation of neighboring countries, might be able to make their own recombinant enzyme preparations for the treatment of Gaucher disease. At first blush, this may seem like a daunting prospect. I have been impressed, however, with the centers of excellence in technology that exist in some countries, even some in which the gross national product per capita is quite low. If the technology for making pharmacologic grade recombinant proteins exists or can be created, this is an option that could be considered. I am not schooled in the law and therefore cannot provide a professional appraisal of the intellectual property landscape. However, as pointed out in the introduction, all of the essential ingredients for the use of enzyme replacement therapy in Gaucher disease are in the public domain. The basic idea was suggested 40 years ago,[1] the first clinical trials were done 30 years ago,[2] the use of mannose targeting was published more than 25 years ago,[8] and recombinant enzyme was made in tissue culture 20 years ago.[44] Thus, I would surmise that the only valid barriers to production of enzyme would deal with details of methods of preparation. Some innovation might be required here. Conclusions The cost of medical care everywhere is rising and no nation can provide everything that healthcare has to offer for everyone. Healthcare budgets are not infinite; what we give to one, we must take away from another. This principle although politically not popular, is a fact of life. It applies to rich nations as well as poor, but it is the poor nations that must make the hard decisions more frequently. Gaucher disease is a disorder that affects very few individuals in the population and its treatment is very costly. Mahatma Gandhi very wisely said "Life is a series of compromises..."(http://www.gandhi-manibhavan.org/gandhicomesalive/l.htm). This holds true for the delivery of healthcare as well as for many other endeavors, and we must learn to compromise in trying to do the best for the most. Acnowledgements This is manuscript number 17894-MEM from The Scripps Research Institute. Supported by the National Institutes of Health grant DK 061370-03 and the Stein Endowment Fund.References
Copyright 2005 - Indian Journal of Human Genetics The following images related to this document are available:Photo images[hg05024f4.jpg] [hg05024f5.jpg] [hg05024f3.jpg] [hg05024f2.jpg] [hg05024f1.jpg] |
| |||||||||

{kind=link}
{kind=link}