 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Human Genetics, Vol. 12, No. 1, January-April, 2006, pp. 26-33 Original Communication Epidemiology and genetics of hypertrophic cardiomyopathy Reena R. Tanjore, Bhavesh Thakkar*, Annapurna D. Sikindlapuram, Calambur Narasimhan*, Prafulla G. Kerkar**, Haragopal V. Vajjha†, Pratibha Nallari Departments of Genetics, Osmania University, *Statistics, Cardiology Unit, King Edward Memorial Hospital, Mumbai, **Cardiology Unit, CARE Hospital, †Department of Statistics, Osmania University, Nampally, Hyderabad, Andhra Pradesh - 500 007, India Code Number: hg06006 Abstract Background: Hypertrophic cardiomyopathy (HCM) is a heart muscle disorder and is known to be inherited as an autosomal dominant trait. Mutations in several sarcomeric, cytoskeletal and mitochondrial genes have been reported in HCM. Though many cases of HCM are being identified, there is limited data regarding the epidemiology and genetics of HCM in India.
Keywords: Hypertrophic cardiomyopathy, inheritance, penetrance, heritability, relative estimates. Introduction Hypertrophic cardiomyopathy (HCM) is a genetically heterogeneous heart muscle disorder characterized by left ventricular hypertrophy, with predominant involvement of interventricular septum in the absence of secondary causes.[1] The prevalence of the disease in the population is reported to be 0.2%.[2] HCM is inherited as an autosomal dominant disorder with variable penetrance in more than 50% of the cases,[3] though sporadic occurrence due to de novo mutations is also observed. Till date mutations in several genes predominantly sarcomeric have been identified to cause HCM, though few nonsarcomeric, cytoskeletal and mitochondrial genes have also been implicated. Although transmission of HCM is usually considered to be dominant, few cases indicating recessive mutations have also been reported.[4],[5] Several cases of HCM in India are beginning to be recognized, however, there is limited data available regarding the true prevelence and epidemiological variables. Further studies on the modes of inheritance in HCM have been carried out more than a decade ago, when only few genes had been identified, but with the emerging molecular advances that had lead to the identification of additional genes causing HCM; this aspect needs to be reassessed. Therefore the present study is envisaged at identifying the epidemiological variables in HCM and fitting a probability model assuming autosomal dominant mode of inheritance based on chi square, segregation, logistic regression analysis and also to assess the penetrance, heritability and relative frequency estimate of the condition, which may give the extent of genetic component involvement and can possibly shed light on the underlying heterogeneity of this complex disorder especially in the Indian context. Materials and Methods From the cardiology unit of CARE hospital, Hyderabad and KEM hospital, Mumbai, 127 patients diagnosed for HCM based on standard clinical, electrocardiographic and echocardiographic criteria were considered for the study. Age, sex, duration of the disease, symptoms, familial status and pedigree information were collected from all the patients. The characteristic feature of HCM is a hypertrophied heart. Based on the pattern of hypertrophy, the HCM cases were classified as asymmetric septal hypertrophy without obstruction (ASH) to flow of blood from the left ventricle, obstructive HCM (HOCM), concentric hypertrophy (Conc HCM), apical HCM, HCM with mid cavity obstruction (MCO) and rare form with hypetrophy in unusual locations, such as posterior portion of the septum and posterobasal free wall. Chi square (χ2) test assuming autosomal dominant mode of inheritance was carried out as reported earlier.[2] Odds ratio (1951)[6] a technique of logistic regression was used to compare whether the probability of the disease is the same for different groups and is given by:
The confidence interval was calculated using the formula Exp {β 1 ± Z 1 -α/2 FNx01 SE (β 1)} where α is the level of significance. The odds ratio can be any nonnegative number. When the row and column variables are independent, the true value of the odds ratio equals 1. An odds ratio greater than 1 indicates that the odds of a positive response are higher in row 1 than in row 2. Values less than 1 indicate the odds of positive response are higher in row 2. The strength of association increases with the deviation from 1. Single′s Incomplete Ascertainment method was followed to calculate the ′probability′ (′p′) value and variance component along with standard error to identify the modes of inheritance (Fischer, 1934). p = R - N / T - N Variance = pq / T - N Where R = Number of affected individuals in all sibships Segregation analysis was carried out on HCM cases to examine for the possible mode of inheritance depending upon the frequency of affected members in the families based on sibship and proportion of affected individuals. Penrose′s method (1953) was adopted to calculate the relative frequencies based on the frequency of the condition in the population ′q′ and sibs ′s′. The mode of inheritance was identified based on the comparison of the observed relative frequencies with those of the expected values, where in the expected relative frequencies were calculated in sibs as 1/2q for an autosomal dominant trait, 1/4q for an autosomal recessive trait and 1/√q for a multifactorial trait and the observed frequencies is given by s/q. The calculated observed probability was examined for its close agreement to that of expected probability estimates of the dominant, recessive or multifactorial modes of inheritance. Based on parental phenotypes and presence and absence of consanguinity the probability of being affected was calculated using Weinberg′s formula to identify heterogeneity among the groups of HCM. Wherein the probability of being affected is given by ∑ r-1 / ∑
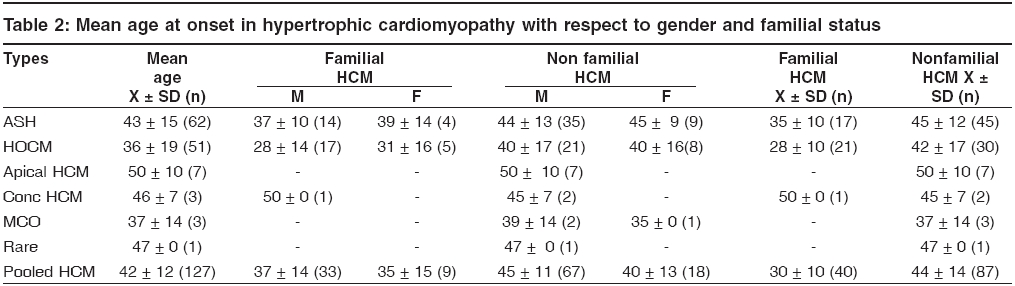
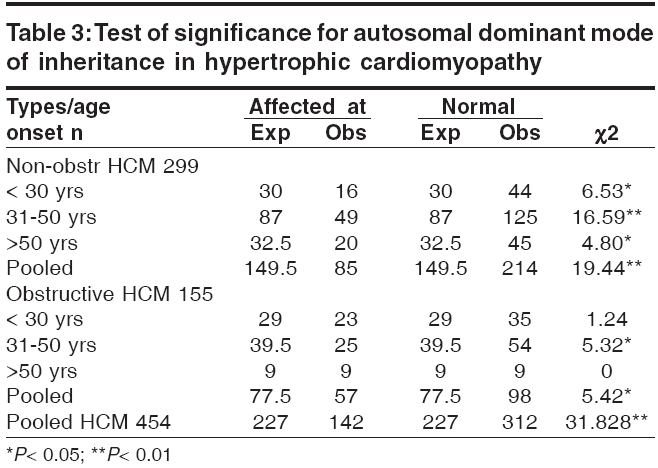
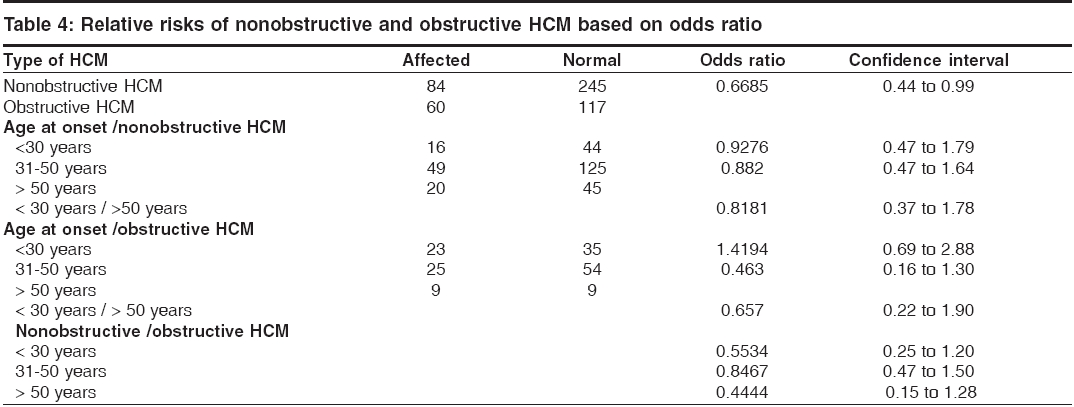
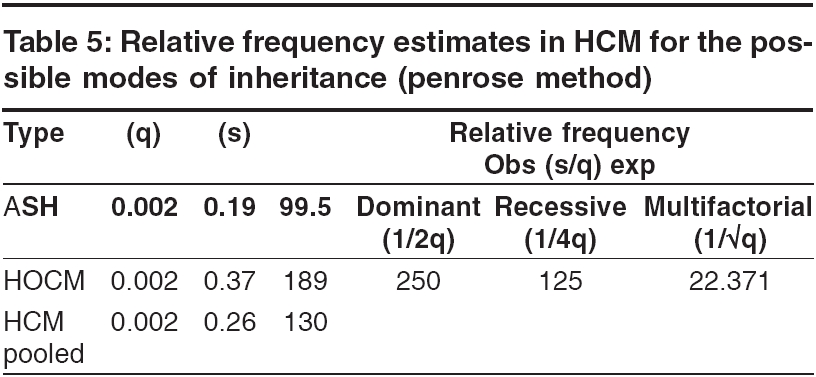
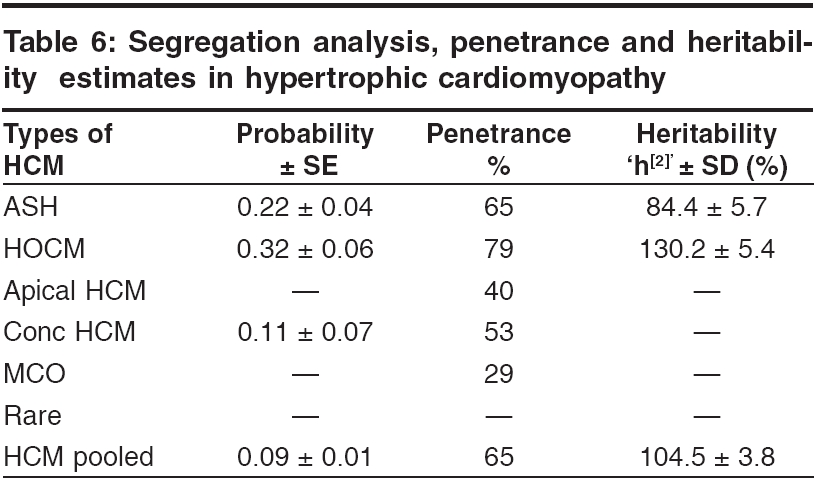
s-1 and To identify the extent of genetic component in the aetiology of HCM, heritability estimates were also carried out by Falconer′s method (1965), in the subsets of HCM and pooled HCM cases. Wherein ′g′ is the frequency of the disorder in general population; ′ra′ is the frequency among relatives; ′x′ is deviation of the threshold from the population mean, ′a′ is deviation of the mean of affecteds from the population mean; ′r′is the correlation between relatives and probands; A = affected individuals in sample; N = total number of individuals. q = frequency = A / N then r = xg - x ra / ag and h2 = r for identical twin and Standard Error h2 = 2√V for first-degree Finally the penetrance of the condition was calculated in percentages based on observed and expected number of affected individuals in 127 sibships, to assess the disease penetrance in affected individuals and their relatives in toto. Results and Discussion [Table - 1] gives the epidemiological variables in HCM cases. In our population the most common pattern of left ventricular hypertrophy was found to be ASH (48%) followed by HOCM (40%) and is in close confirmation with other studies reported elsewhere.[7],[8] Apical HCM constitutes 25% of cases in Japan but only 8-10% cases in the non-Japanese population,[9],[10] whereas our study revealed only 5.5% of apical HCM cases. Further only 2% cases of concentric HCM were observed, which is low compared to other reports of 20-25%.[7],[8] Mid-ventricular obstruction was seen in 2% of the patients apart from one case of rare form of HCM involving mid and distal interventricular septum and apex. The subtypes of HCM clearly reveal the heterogeneous nature of the condition. Our study showed an increased preponderance of the condition among the males (78.7%) when compared to females (21.3%), with the sex ratio being 3.7:1, which indicates a higher male preponderance compared to previous studies from western (2.9:1) and Japanese (2.3:1) populations.[11],[12] The sex ratio in the different subsets of HCM were found to be 3.7:1 for ASH, 2.9:1 for HOCM, 2:1 for HCM with mid cavity obstruction. The male preponderance could be attributed to various factors like developmental, anatomical, hormonal and environmental variations. These differences probably confer greater vulnerability to hypertrophic stimuli, left ventricular wall stress, diastolic and systolic dysfunctions in males. Alternatively, females in general have blunted cardiac responses compared to their male counterparts following manipulation of a number of genes and also endogeneous estrogen may delay the onset of symptoms in females by several cardiovascular protective mechanisms. Hence studying these differences could shed light on the multifaceted mechanisms involved in the aetiopathogenesis of HCM. In general 31% of the HCM probands have a familial history of the disease, which is in conformation with other reports.[13] Further HOCM was found to have a higher familial status (41%) compared to ASH (27%), while 66% (2 out of 3 cases) individuals with concentric HCM had a family history. None of the patients with apical, mid cavity obstruction and rare form of HCM had family history and the familial status in these types needs to be confirmed in a large sample. The differences in the familial status observed, could be attributed to the genetic heterogeneity of the different subsets of HCM. Sudden death, the most serious complication of HCM, may occur as the presenting manifestation of disease or at any time throughout the course of disease. Of the total 95 cases, 3 (~3%) probands expired due to HCM related complications during a two-year follow up study. Previous studies on sudden cardiac deaths in HCM patients report an annual mortality of 2-6% with 21% having a family history of sudden death.[14],[15] However, the family history of sudden death in our study was found to be 27.5% for pooled HCM, 37% for HOCM, 22.5% for ASH and 66% for concentric HCM. Decreased availability of health care along with under diagnosis may explain such a higher rate of death in our population. The observation that history of sudden death was twice the frequency among obstructive HCM cases as compared to non-obstructive form, indicates the malignant nature of obstructive HCM associated with poor prognosis. [Table - 2] gives the mean age at onset in HCM with respect to gender and familial status. The expression of HCM is usually age related, occurring during or soon after periods of rapid somatic growth. The mean age at onset in general was found to be 42 ± 12 yrs and among the different subsets of HCM, the onset of the disease was early in HOCM (36 ± 19yrs) and in MCO (37 ± 14 yrs), as compared to the other forms: 43 ± 15 yrs for ASH, 50 ± 10 yrs for apical HCM and 46 ± 7yrs for concentric HCM. In an individual who was identified with the rare form the age at onset was 47 yrs, clearly indicating HCM to be a 3rd and 4th decade disorder. Inter group comparisons based on the familial status revealed that the age at onset was more than a decade early in the individuals with a family history of HCM (30 ± 10 yrs) compared to non- familial cases (44 ± 14 yrs). This was observed even among the subsets of HCM, wherein for ASH the age at onset for familial cases was 35 ± 10 yrs compared to non familial cases (45 ± 12 yrs), in familial HOCM the onset was 28 ± 10 yrs compared to 42 ± 17 yrs for non familial cases. The findings clearly highlight that familial cases are genetic in nature and anticipation of the HCM can be predicted in unaffected family members and/or their progeny. In concentric HCM cases, the age at onset for non-familial cases was 45 ± 7 yrs and only one individual (50yrs) reported with a family history. Owing to the small sample size and absence of familial cases, inter group comparisons were not possible for apical, mid cavity obstruction and rare forms of HCM. A study by Jung et al , (2000)[16] on familial and non-familial HCM cases suggests that the severity of metabolite abnormalities is different in familial and non-familial cases. This was concluded from the significantly increased Pi/PCr ratio (Pi-inorganic phosphate; PCr-phosphocreatine) and from trends towards a greater PME/PCr (PME- phosphomonoesterase) and a smaller PCr/ATP ratio in familial HCM though no obvious difference in the extent of hypertrophy was observed when compared to non-familial cases. Thus individuals with a family history showed a strikingly different metabolite ratio in their myocardium, which is in relation to their inherited gene mutation and hence, they may present early with symptoms due to their genetic predisposition. Inter group comparisons based on gender in the various subsets of HCM showed the difference to be ≤ 5 yrs, implying that the age at onset for HCM is influenced more by the familial status and type of HCM rather than sex of an individual. The assumption for autosomal dominant mode of inheritance was carried out by chi square (χ 2sub) analysis [Table - 3]. The different subsets of HCM were grouped into two classes a) non-obstructive HCM including ASH, apical, concentric and rare form of HCM b) obstructive HCM including HOCM and HCM with mid cavity obstruction. Chi square tests revealed that both non-obstructive (χ2 =19.44; P < 0.05) and obstructive HCM (χ2 =5.42; P < 0.05) deviated significantly from the said autosomal dominant mode of inheritance, but the magnitude of deviation in the case of non-obstructive HCM was higher as compared to obstructive HCM. Further, at higher level of significance ( P < 0.01) obstructive HCM was following autosomal dominant mode of inheritance whereas non-obstructive HCM was significantly deviating. The deviation observed may be attributed to incomplete penetrance of the condition in case of obstructive HCM, this alone may not justify the deviation in case of non-obstructive HCM cases, implying a different mode of inheritance. The sibships were further subclassifed based on age at onset into three groups: early onset (< 30 yrs), middle onset (31-50 yrs) and late onset (>50 yrs) and were tested for the possible mode of inheritance. In the non-obstructive class, all the three groups i.e early (χ2 =6.53; P < 0.05), middle (χ2 =16.59; P < 0.05) and late onset (χ2 =4.80; P < 0.05) groups, deviated significantly from the autosomal dominant inheritance pattern. In the obstructive HCM class the early (χ2 =1.24; P < 0.05) and late onset (χ2 =0) groups followed autosomal dominant inheritance pattern, whereas the middle onset group (χ2 =5.32; P < 0.05) deviated significantly. In both the classes of HCM, the level of deviation is significantly high for the middle onset group as compared to early and late onset groups of HCM and therefore this group can be considered as an admixture wherein genes/gene modifiers and environmental variables may be contributing to the heterogeneity. This is further supported by familial status and molecular studies of different gene mutations in which it was found that β -myosin heavy chain,α-tropomyosin and troponin T mutations are usually associated with early onset of the disease, where as myosin binding protein C, troponin I and a-myosin heavy chain mutations were identified in the late onset HCM cases.[17],[18],[19],[20],[21] Hence, molecular studies keeping in view of the age at onset as a criterion, needs to be addressed on a large sample data. [Table - 4] gives the relative risk of non obstructive and obstructive HCM based on odds ratio. The odds ratio was found to be 0.66 for non obstructive and obstructive HCM comparisons indicating that the proportion of affected individuals is higher in the obstructive group further strengthening the earlier observation of dominant mode of inheritance with high penetrance and high familial nature. Further intra group comparisons in the non obstructive HCM group based on age at onset showed the odds ratio to be 0.92 for early and middle onset group, 0.82 for middle and late onset and 0.81 for early and late onset group comparisons respectively. The odds ratio in the above cases is close to 1 indicating that in the non obstructive HCM group there is no variation in the risk to HCM with respect to age at onset and that some other epidemiological variables may be associated. Similar comparision in the obstructive HCM group showed the odds ratio to be 1.41 for early and middle onset group, 0.46 for middle and late onset group and 0.65 for early and late onset groups respectively. The odds ratio are significant in this group indicating high gene penetrance and familial status in the early and late onset groups when compared to middle onset group of obstructive HCM. Finally inter group comparisons between non obstructive and obstructive HCM based on onset showed significant association in early (0.55) and late onset (0.44) groups of obstructive HCM, with the risk being more or less similar (0.84) in the middle onset groups for both non obstructive and obstructive HCM. Thus heterogeneity with respect to age at onset is observed in obstructive HCM also suggesting that middle onset group is more likely to be under the influence of stress and environmental factors. [Table - 5] gives the relative frequency estimates for the possible modes of inheritance in HCM. The Penrose relative frequency estimates were carried out to establish the familial concentration by comparing the frequency of the condition in the general population and among relatives of the affected proband to check for the possible modes of inheritance in HCM. Since, the frequency of the condition in the general population is reported to be 1 in 500, the ′q′ value was taken as 0.002 and the frequency of the condition among the sibs, ′s′was found to be 0.19 for ASH and 0.37 for HOCM. The observed number of affected individuals based on relative frequency estimates was found to be 99.5 for ASH, 189 for HOCM and 130 for pooled HCM cases. Due to small sample size and absence of family history, the other subsets of HCM could not be included in the present analysis. The expected number of affected individuals was 250 assuming autosomal dominant inheritance, 125 for autosomal recessive and 22.3 for multifactorial mode of inheritance respectively. The relative frequency estimates support the autosomal dominant inheritance pattern with reduced penetrance for HOCM whereas ASH seems to be more closely following recessive mode of inheritance. Hence, HCM can be considered to be a mixed model inheritance based condition. [Table - 6] gives the segregation analysis, penetrance and heritability estimates in hypertrophic cardiomyopathy. The probability estimates based on sibships and incomplete ascertainment was carried out for the conformation of the dominant mode of inheritance as reported earlier in HCM. The ′ P ′ value and standard error calculated by Single′s Incomplete ascertainment method were found to be 0.22 ± 0.04 for asymmetric septal hypertrophy, 0.32 ± 0.06 for obstructive HCM, 0.11 ± 0.07 for concentric HCM and 0.09 ± 0.01 for pooled HCM against the expected value of 0.5 for autosomal dominant disorders. The overall penetrance estimates for pooled HCM and ASH was found to be 65%, 79% for HOCM, 40% for apical HCM, 53% for concentric HCM and 29% for HCM with mid cavity obstruction. Thus the highest penetrance was observed for HOCM, followed by ASH, while apical and concentric HCM showed moderate penetrance, the lowest penetrance was observed in mid cavity obstruction subset. Reduced penetrance and variable manifestation of the condition implies that other factors (both genetical and environmental) affect the expression of the disease. The variation observed in the ′ P ′values and variable penetrance among the subsets of HCM further strengthens the underlying genetic heterogeneity of the condition and pinpoints towards the involvement of different genes/mutations (functional domains). Hence, modes of inheritance other than dominant may be encountered, in ASH, apical, concentric and mid cavity obstruction types. Alternatively these subsets of HCM could be more influenced by environmental and gene modifiers compared to ASH and HOCM and hence a mixed model of inheritance is a best fit for such complex disorders. Heritability estimate is the most common statistic in quantitative genetics for expressing the importance of transmissible genetical effects. The heritability estimates was found to be 84 ± 5.7 for ASH and 130 ± 5.4 for HOCM. In general, the heritability of liability estimates are high, confirming the strong familial nature of the condition and indicating greater involvement of genetic component in HCM aetiology. Estimates over 100% as observed in HOCM and pooled HCM, could be attributed to autosomal dominant mode of inheritance and and/or major loci segregating for the condition, whereas non obstructive cases, revealed an estimate of 84% signifying the genetic and environmental/gene modifier interactions. The probability of an individual being affected in a family was calculated by Weinberg′s formula, taking into consideration the parental phenotypes and presence or absence of parental consanguinity [Table - 7]. Low parental consanguinity was observed in general, irrespective of the parental phenotypes. The probability estimates were 0.272 and 0.230 when both the parents were phenotypically normal, whereas when one of the parent was affected the estimates were 0.392 and 0.333, clearly pinpointing the inter group variation implying the genetic heterogeneity of the condition and focusing on the autosomal dominant and recessive modes of inheritance. Conclusions The epidemiological findings of the present study can be considered as preliminary and first of its kind from our population. The subtypes of HCM reveal the heterogeneous nature of the condition with the influence of genotypes on their morphology. Distinctive differences in the sex ratio, familial status, family history of sudden cardiac death and variable age at onset among the subsets of HCM implicate the involvement of different genes / mutations apart from the differential influence of gene modifiers/environmental effects. Thus the study could bring out the complexity associated with HCM and suggests the mixed inheritance model from the Indian context, which is not in conjunction with earlier reports. The earlier reports support autosomal dominant mode of inheritance in case of familial HCM, whereas the present study proposes that other modes of inheritance like autosomal recessive may be encountered in a subset of non-obstructive HCM. Hence attempts to unfold the specific molecular pathways for this multifaceted complex disease with more refined geno and phenotyping may enable us to unravel unsuspected physiological mechanisms and modifier genes of direct relevance to HCM.References
Copyright 2006 - Indian Journal of Human Genetics The following images related to this document are available:Photo images[hg06006t7.jpg] [hg06006t6.jpg] [hg06006t4.jpg] [hg06006t2.jpg] [hg06006t5.jpg] [hg06006t1.jpg] [hg06006t3.jpg] |
| |||||||||

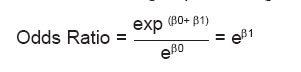
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}