 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
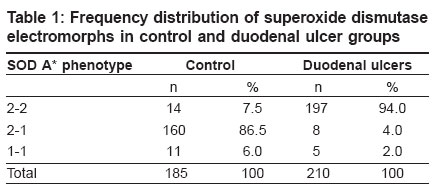
Indian Journal of Human Genetics, Vol. 12, No. 3, September-December, 2006, pp. 125-128 Original Communications Superoxide dismutase phenotypes in duodenal ulcers: A genetic marker? Sulekha S, Madhavi J, Venkateshwari A, Yasmeen S, Pratibha N Department of Genetics, Osmania University, Hyderabad - 500 007 Code Number: hg06023 Abstract Background:Cu-Zn superoxide dismutases are antioxidative defensive enzymes that catalyze the reduction of superoxide anions to hydrogen peroxide.
Aim:The study focuses on the association of electromorph of superoxide dismutase with duodenal ulcers, which result due to an imbalance between aggressive and defensive factors.
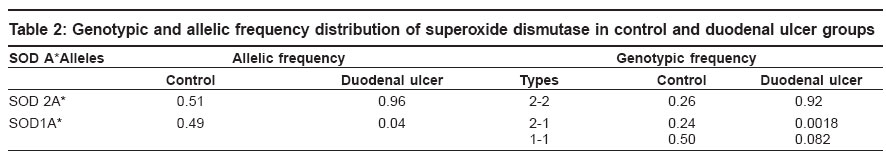
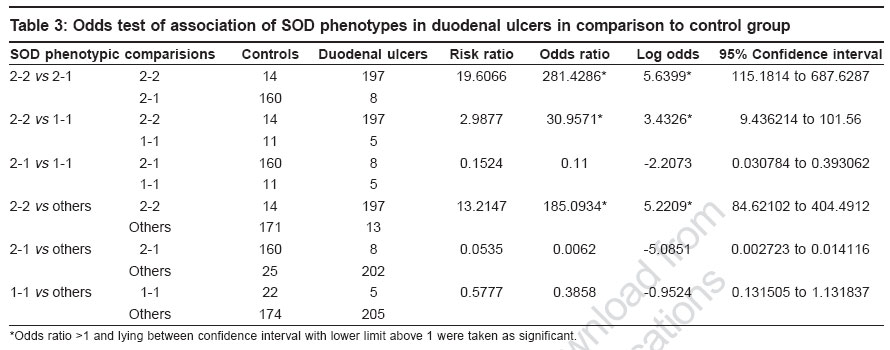
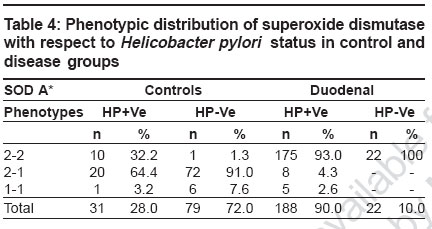
Keywords: Antioxidant, duodenal ulcer, free radical, gene modifier, genetic marker, lipid peroxidation, superoxide dismutase Introduction Duodenal ulcers (DU) constituting the predominant form of peptic ulcers, found to occur in about 10% of the worldwide population, are characterized by the loss of mucosal resistance, as a result of action and interaction of several aggressive factors like Helicobacter pylori infection, acid, pepsin and other proteases secreted on one hand and several defensive factors like bicarbonate and antiprotease on the other hand. Oxidative stress also plays an important role in vascular physiology and pathology.[1] Previous studies have shown that superoxide anion is produced by oxidases present in human vascular cells.[2] However, net levels of oxidative stress are determined in parallel by production and biodegradation of free radicals as well as by genetic susceptibility. The major anti-oxidant systems in the mucosal vasculature include, catalase, glutathione peroxidase, thioredoxin and superoxide dismutases, which modulate the oxidative stress but the primary role in the metabolism of superoxide anion radical is exerted by superoxide dismutases.[2] Superoxide dismutases (SOD) is well known for scavenging Superoxide radicals such as reactive oxygen species (ROS), subsequently protecting cells from oxidative injury and for maintaining tissue homeostasis. SOD is polymorphic in nature with three identifiable phenotypes SOD A*1, SOD A*2-1 and SOD A*2 encoded by two allelic forms SOD A*1 and SOD A*2 respectively and inherited as co-dominant alleles.[3] The gene is localized to 21q21-q22.1 region.[4] Since no information with regard to SOD and its electromorph association is available with respect to duodenal ulcers, such a study is imperative to identify the role of susceptible marker and its associated allele, apart from its role as a modifier gene in the genetic etiology of duodenal ulcers. Therefore, the present study aims at identifying specific electromorph association of SOD in duodenal ulcers with respect to Helicobacter pylori and its possible role in the etiopathogenesis of duodenal ulcers. Materials and Methods Blood samples from endoscopically confirmed cases comprising of 210 duodenal ulcers and 185 healthy controls matched for age and sex, were collected from Department of Gastroenterology, Osmania General Hospital , Hyderabad and voluntary blood donor camps, Hyderabad. Five ml of venous blood was collected in EDTA to obtain red blood cell membrane. The whole blood was washed with normal saline thrice and then centrifuged with ice-cold water to obtain red cell membranes for the phenotyping of superoxide dismutase. The Helicobacter pylori infection status was tested by rapid urease test in mucosal tissue biopsies of the duodenal ulcers as described by Vaira et al.[5] The phenotyping of SOD was based on Davies protocol[6] of an 8% polyacrylamide gel electrophoresis gel and the electrophoresis was carried out using 0.005 M tris glycine running buffer at 4°C for two hours at 20 mA constant current. The gel was then placed in a tray and incubated in freshly prepared staining solution which consisted of 5 ml of phosphate buffer (pH 7.8), 8.5 ml of riboflavin, 10 mg of nitroblue tetrazolium chloride and 220 μl of TEMED prepared in 50 ml distilled water. Later, the gel was washed and transferred to distilled water and exposed under a bright fluorescent light till the background was blue and clear achromatic bands were visualized as cited by Beauchamp and Fridovich.[7] Based on their mobility in the gel, the phenotypes of SOD A were identified as 2-2, 2-1 and 1-1, respectively. Frequency distribution was computed and allelic frequencies of SOD phenotypes were estimated to test for the Hardy Weinberg equilibrium[8] and odds ratio (2 x 2 contingency) at 95% confidence interval limits was carried out to interpret the results statistically.[9] Results [Table - 1] shows the frequency distribution of superoxide dismutase electromorphs in control and disease groups wherein SOD 2-1 phenotypes were found to be predominant in normal individuals (86%) while 2-2 phenotypic individuals are at an increased risk to the disease condition (94%). The distribution of superoxide dismutase genotypic and allelic frequencies in control and duodenal ulcer groups is shown in [Table - 2]. The frequency of SOD 2 and SOD 1 alleles were found to be 0.55 and 0.45 in healthy subjects and 0.96 and 0.04 in duodenal ulcer patients. A significant deviation from the Hardy- Weinberg equilibrium was observed in duodenal ulcer group strengthening the earlier observation [Table - 1]. The odds ratio estimates of superoxide dismutase phenotypes in duodenal ulcer groups in comparison to control group, wherein an increased risk of SOD 2-2 phenotypic individuals to ulceration (log OR 5.6399) was observed, further strengthening the above observation [Table - 3]. This could be attributed to SOD A*2 allele encoding for a less stable enzyme as a result of which altered scavenging of the superoxide free radicals may lead to oxidative stress induced mucosal damage, as seen in duodenal ulcers. Decreased activity of Cu-Zn SOD is also associated with initiation of nitric oxide (NO-) mediated apoptotic cell death and hence tissue damage in ulcers groups.[10] [Table - 4] shows the phenotypic distribution of SOD with respect to Helicobacter pylori status in control and disease patients. In general, an interesting observation made is the predominance of SOD 2-2 phenotype in Hp +ve healthy subjects and ulcer patients, but the nature of association cannot be demonstrated statistically as the frequency distribution of 2-1 and 1-1 phenotypes in Hp-ve cases accounted to zero, further supporting the role of SOD as a gene modifier in such Helicobacter pylori infected cases. However, by observation, role of SOD A* 2 allele scavenging the O 2- derived free radical released due to mucosal damage as a consequence of Helicobacter pylori infection further justifies the SOD as a genetic marker as well as modifier of the disease in such Hp +ve cases. The study thus substantiates the role of SOD in the etiopathogenesis of duodenal ulcers and helps in genetic heterogeneity and risk prediction of duodenal ulcer groups. Discussion Cells have elaborated protective mechanisms to cope with potentially damaging molecules such as reactive oxygen species by synthesizing antioxidant enzymes.[10] Superoxide dismutases (SOD) are key cellular defense systems that disproportionate O 2- into oxygen and H 2 O 2 , with the latter being detoxified by glutathione peroxidase or catalase. Cu-Zn SOD referred to as SOD 1 and Mn SOD as SOD 2 are the two eukaryotic SOD′s. Cu-Zn SOD located in the cytosol, is expressed constitutively and is considered as a housekeeping enzyme; in contrast to Mn SOD which is located in the mitochondrion. The Cu-Zn SOD enzyme is a known polymorphic marker encoded by two allelic forms SOD A*1 and SOD A*2 and inherited as co dominant alleles. An increased risk of SOD 2-2 phenotypic individuals to ulceration (OR: 281.43* CI: 115.18 to 687.68) was observed, this could be explained on the basis of SOD A*2 allele contributing to decreased stability of the enzyme or lowered enzyme synthesis. Studies by Novak et al[11] revealed significant reduction in SOD activity and hence the locus might be ′regulatory′ in nature wherein the altered structure and function of cell membranes especially of gastric mucosa as a result of ulceration, may have a key pathogenetic role in ulcers. SOD, a key enzyme in gastric mucosal protection, is depleted significantly in the ulcer edge compared to the controls and found to increase after healing with treatment.[12] Hence, significant association of the SOD A*2 allele encoding for less stable enzyme, as a gene modifier in duodenal ulcers is justifiable. Further, SOD A*2 allele contributing to less stable enzyme can have direct effect on lipid peroxidation and hence pathological progression of the disease. Significant elevation of malondialdehyde (MDA) in duodenal ulcers was reported which strengthens the oxidative stress mechanism associated with duodenal ulcers.[13] Since MDA levels are negatively correlated any altered function of SOD enzyme may in turn lead to increased MDA levels, disrupting the homeostatic balance causing high lipid peroxidation of the membranes of gastric mucosa[14],[15] which may culminate to ulceration, due to depletion of SOD levels. Previous studies suggest that only Mn-SOD plays an important role as an antioxidant against ROS and, sub-sequently, in the maintenance of cell turnover in gastric mucosa.[16] Gotz et al have shown that gastric tissue of Helicobacter pylori positive individuals was higher in activity and content of the cytokine inducible Mn-SOD, with no change in the constitutive Cu-Zn SOD.[17] Hence Cu-Zn SOD role is rather trivial in case of Helicobacter pylori infection in duodenal ulcers. Our results are strengthened by the above observation implicating no association of Helicobacter pylori infection and Cu-Zn SOD enzyme. Conclusion The study highlights a significant risk of homozygous SOD A*2-2 phenotypic individuals to duodenal ulcers and SOD A*2 allele encoding for a less stable enzyme may lead to decreased scavenging of O 2- radicals resulting in the oxidative stress induced gastric mucosal tissue damage and ulcerogenesis. Thus SOD may be considered as a genetic marker in genetic risk prediction and may have a direct or modifier role in ulcerogenesis and could help in delineating the genetic heterogeneity of duodenal ulcers. References
Copyright 2006 - Indian Journal of Human Genetics The following images related to this document are available:Photo images[hg06023t3.jpg] [hg06023t1.jpg] [hg06023t2.jpg] [hg06023t4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}