 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
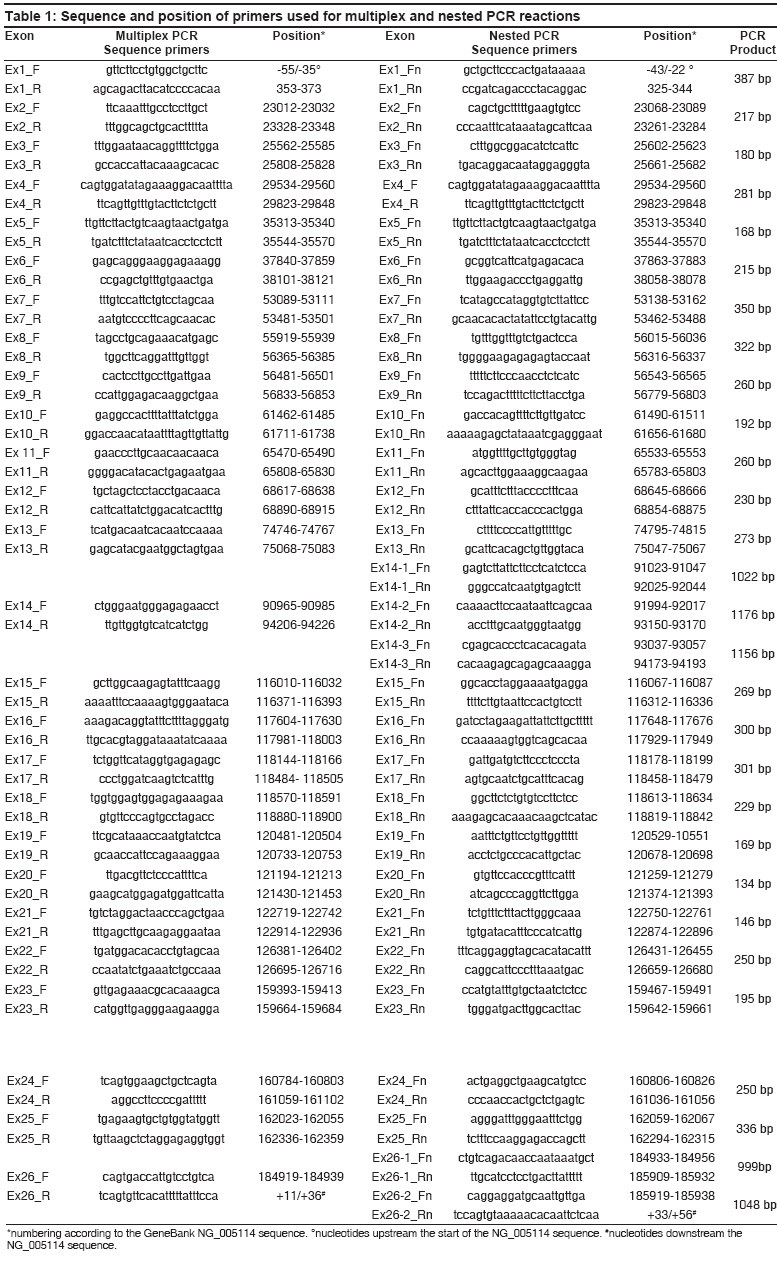
Indian Journal of Human Genetics, Vol. 14, No. 2, May-August, 2008, pp. 55-64 Original Article Identification of FVIII gene mutations in patients with hemophilia A using new combinatorial sequencing by hybridization Chetta M, Drmanac A, Santacroce R, Grandone E, Surrey S, Fortina P, Margaglione M Genetica Medica, Dipartimento di Science Biomediche, Universita' degli Studi di Foggia, Foggia; Callida Genomics, Sunnyvale, CA Code Number: hg08013 Abstract Background: Standard methods of mutation detection are time consuming in Hemophilia A (HA) rendering their application unavailable in some analysis such as prenatal diagnosis.Objectives: To evaluate the feasibility of combinatorial sequencing-by-hybridization (cSBH) as an alternative and reliable tool for mutation detection in FVIII gene. Patients/Methods: We have applied a new method of cSBH that uses two different colors for detection of multiple point mutations in the FVIII gene. The 26 exons encompassing the HA gene were analyzed in 7 newly diagnosed Italian patients and in 19 previously characterized individuals with FVIII deficiency. Results: Data show that, when solution-phase TAMRA and QUASAR labeled 5-mer oligonucleotide sets mixed with unlabeled target PCR templates are co-hybridized in the presence of DNA ligase to universal 6-mer oligonucleotide probe-based arrays, a number of mutations can be successfully detected. The technique was reliable also in identifying a mutant FVIII allele in an obligate heterozygote. A novel missense mutation (Leu1843Thr) in exon 16 and three novel neutral polymorphisms are presented with an updated protocol for 2-color cSBH. Conclusions: cSBH is a reliable tool for mutation detection in FVIII gene and may represent a complementary method for the genetic screening of HA patients. Keywords: Combinatorial sequencing-by-hybridization, FVIII gene, hemophilia A Introduction Hemophilia A (HA) is a common inherited recessive X-linked disorder of blood clotting caused by deficiency of factor VIII in the coagulation cascade and affects approximately 1 in 5,000 males world-wide. [1],[2] The FVIII gene, comprises 26 exons ranging from 69 bp (exon 5) to 3.1 kb (exon 14) in size, spans 186 kb of genomic DNA and produces a 9030 nt mRNA. According to the UK Hemophilia Centre Doctors′ Organisation (UKHCDO) Hemophilia Genetics Laboratory Network, the severity of HA in the pedigree should be determined first as this will influence the diagnostic strategy to be employed. Severe Hemophiliacs should be screened for the intron 22 inversion mutation followed by the intron 1 inversion mutation. This approach identifies the underlying mutation in 45-50% of severe HA patients. [3],[4] The remaining severe HA pedigrees should then be analyzed further either by full mutation or linkage analysis. Mutations have been found in nearly all 26 exons of the factor VIII gene, over 400 mutations have been identified [5],[6] and de novo mutations represent approximately 30% of all cases. [7] The most common detection methods include DNA sequence analysis which requires numerous reactions and individual analysis of each exon or alternative screening methods such as single-stranded conformation polymorphism (SSCP), [8] denaturing gradient gel electrophoresis (DGGE) [8] and denaturing high performance liquid chromatography (dHPLC). [9] We applied the new combinatorial sequencing-by-hybridization (cSBH) as an alternative method to the traditional Sanger dideoxy chain termination approach. [10],[11] Previous works have shown that cSBH is an efficient rapid and alternative method for mutation detection. [12],[13] We increased the quality of results with a new cSBH method that use two different colors (TAMRA and QUASAR). The platform is an indirect method which uses standard chemistry of base-specific hybridization of complementary nucleic acids to indirectly assemble the order of bases in a target DNA. Short oligonucleotide probes are arrayed in the form of high-density arrays of universal sequence and hybridized to sample DNA molecules. The resulting hybridization pattern is used to generate the target sequence using computer algorithms. We report development of a strategy to implement 2-color cSBH to screen a range of mutations within the FVIII gene. Materials and Methods Sample Primer design DNA amplification The nested PCR amplification for all exons was performed in 100 µ l reactions containing a 1:4 dilution of multiplex PCR reaction (25 µ l multiplex reaction in 75 µ l of ddH 2 0), 30 pmol of forward primer and 3 pmol of reverse primer, 0.6 U of polymerase (England Biolabs) to amplify all exons except exons 14 and 26 that required 1 U of polymerase (England Biolabs), and 1X PCR Callida′s buffer from Callida′s Genomics. Exon 14 (3.1 kb) and 26 (1.9 kb) required three and two overlapping nested asymmetric PCR, respectively. All PCR reactions were performed using Gene Amp PCR system 9700. PCR reactions were for 45 cycles with an initial denaturation at 96°C and for 3 min, followed by 96°C for 30 sec, annealing at 55°C for 30 sec and extension at 72°C for 1 min. Final extension was at 72°C for 1 min. An aliquot of the PCR product was analyzed on a 1% (w/v) agarose gel to verify size and quality of the PCR product. The single strands are detected with gel-star Nucleic acid Gel stain (Cambrex, Rockland, UK). Probes Target preparation Precipitated DNA was resuspended in 40 µ l of dH 2 O and therefore, the double-stranded PCR template was eliminated by restriction digestion leaving single strands intact. The digestion was performed using Callida′s pre-made frozen mix in a total volume of 50 µ l with an optimized amount of DNase I (Gibco-BRL, Rockville, MD) at 37°C for 15-20 min followed by inactivation at 95°C for 5 min. Hybridization The volume of 120 µ l of DNA-ligase mix (27 µ l) was dispensed into each of the eight labelled 5-mer TAMRA and QUASAR linked probe pools pre-aliquoted in a strip of eight small tubes supplied by Callida. Eight probe pools containing target DNA and ligase were mixed thoroughly and 27 µ l was loaded by pipette onto the HyChip cartridge, where it was drawn by capillary forces into the selected hybridization chamber. Hybridization and ligation occurred in a humidity chamber at room temperature for 60 min. Slides then were hot-washed (60-65°C) to remove non-ligated labelled probes using Callida′s detergent (containing washing buffer) for 15 min in an orbital shaker 150 rpm, rinsed four times in dH 2 O and spin-dried at 1500 rpm for 3 min. Slides then were scanned at 20 µ m pixel resolution using a standard array reader GenePix ® 4000B setting PMT (532 nm) = 650 and PMT (635 nm) = 750. Analysis
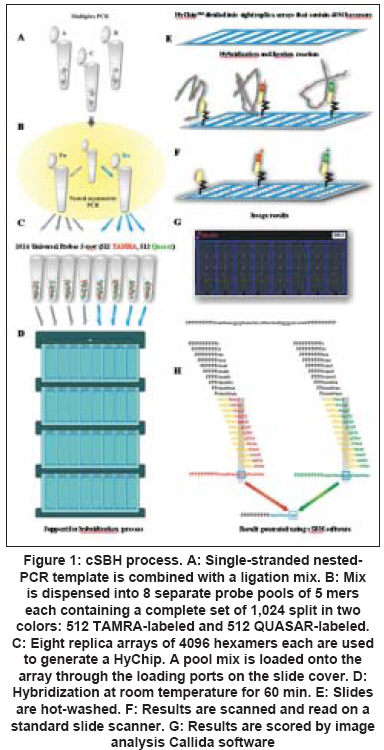
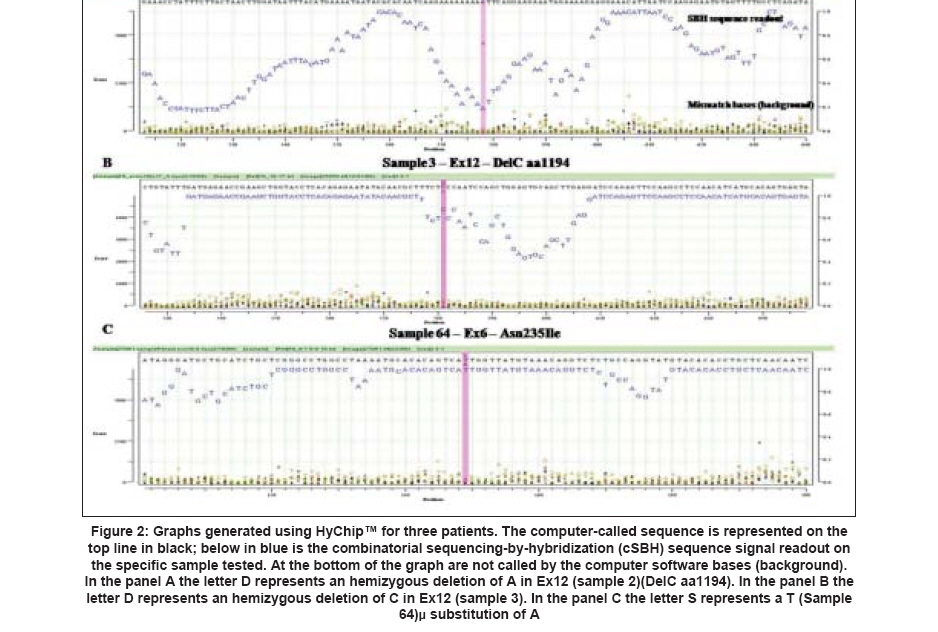
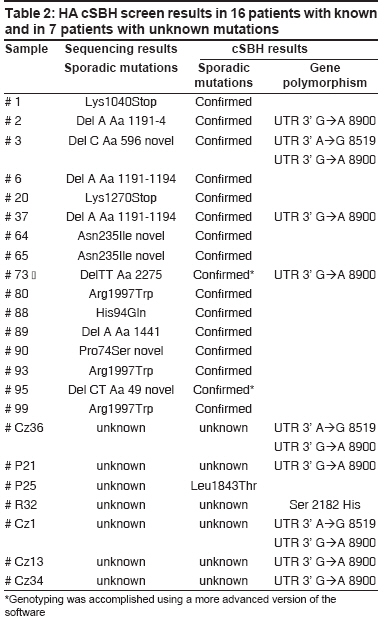
Results The new 2-color cSBH is an indirect sequencing method in which overlapping probes of known sequence are hybridized to DNA target molecules. In cSBH, which is represented in a schematic overview in [Figure - 1], target DNA from a PCR product is exposed to two universal sets of short probes in the presence of DNA ligase. When both array-bound and solution-phase labelled probe hybridize to target DNA at precisely adjacent complementary positions, they are covalently linked by DNA ligase, creating one long, array-bound labeled probe of known sequence that is complementary to the solution-phase, single-stranded PCR-generated DNA template. A schematic presentation of a cSBH base-calling chromatogram is shown in [Figure - 2]. The y-axis represents the relative intensity score of the hybridization signal of all overlapping probes at each base. In the presence of strong hybridization signals, the SBH readout appears high, and the reverse is true when there are weak probe hybridization signals. The x-axis represents the position of each base in the sequence analyzed. The computer-called sequence, compared to the known reference sequence used, is represented as the top line of the sequence (in black), and the actual SBH sample sequence readout is directly below (in blue). Background bases, which are not called by the computer software, are present at the bottom of all graphs. Use of informative pools of labelled probes allows combinatorial ligation of all labelled and bound probes on one glass slide with only eight universal replica arrays of bound probes. This is the most efficient way to obtain hybridization data for all 4,194,304 11-mers using 32,768 (8 X 4,096) probe spots, an array that can fit only one half of all possible 8-mers. Data of pooled 11-mers are much more informative than data for individual 8-mers because each base is read with 11 instead of eight overlapping probes. Mutation Detection in Italian Haemophilic A Patients A total of 16 clinically diagnosed Italian haemophilic A patients and one obligate carrier previously genotyped and found to have disease-causing mutations in the FVIII gene were included in this study. Patient samples included a range of DNA mutations mapping throughout the FVIII gene. cSBH results are shown in [Table - 2]. We confirmed single-base changes in samples 1, 20, 64, 65, 80, 88, 90, 93, 99. Previous genotyping of samples 2 [Figure 2A], 3 [Figure 2B], 37, and 89 carrying a single-base deletion in exon 14 (fragment b) as well as in sample 6 [Figure 2C] containing a single deletion in exon 12 was confirmed by cSBH. In addition, we found polymorphism in the 3′UTR region (Ex 26, nucleotide 8900 (G→A)) in sample 2, 3, 37, 73, and another polymorphism in the 3′UTR region at nucleotide 8519 A→G in sample 3. The correct genotyping of a patient (sample 95) and an obligate carrier (sample 73) with 2-bp deletions starting at codons 2275 and 49, respectively, was accomplished using a more advanced version of the software. We also analyzed samples of 7 haemophilic patients with unknown mutations, who were excluded to carry one of the common HA mutations, inversion of the intron 22 or intron 1. In this setting, we have found a previously undetected missense mutation Leu1843Thr in exon 16 in the sample P25. The mutation was then confirmed by direct gene sequencing. In sample R32 we found another polymorphic change in Ex 23 (Ser 2182 His). In addition, we found a polymorphism in the 3′UTR region (Ex 26, nucleotide 8900 (G→A)) in sample P21, Cz1,Cz13 and Cz34 and Cz36 and another polymorphism in the 3′UTR region at nucleotide 8519 AàG (samples Cz 36 and Cz 1). Discussion We demonstrated that cSBH, and the related HyChip product, is capable of identifying a range of point mutations located within the FVIII gene making this platform an attractive alternative to standard sequencing methods. We confirmed known FVIII gene mutations and found that large regions of genomic DNA can be sequenced with excellent readability using only one HyChip array. Because the maximal length of a single PCR product that could be sequenced by the chip is 1.2 kb in size, long exons such as exons 14 and 26 had to be divided in three and two fragments, respectively. The other exons are shorter and could be sequenced in entirety. cSBH sequence data showed 100% accuracy with only 0.2% of bases not called. [10] In addition, we demonstrated that the smaller exons 1-25 could be pooled together in groups in a manner allowing their complete sequencing on four different chips, with base readability of 100% and an accuracy of 100%. The reliability of this technique has been further provided by analyzing a series of HA patients in whom no gene mutation was detected by the first FVIII gene screening, using common strategies (SSCP, DGGE, or dHPLC). This approach was able to identify a new point mutation (Leu1843Thr), which was subsequently confirmed by direct gene sequencing. In the remaining patients, no mutation was found also when the direct gene sequencing approach was applied. Finally, how well this technique does work was provided by the identification of the mutant allele in a heterozygous carrier with one normal factor VIII gene. Recently, a different approach using a microarray technology has been applied for the detection of FVIII gene mutations. [14] As compared with that, the method we used was able to screen all exons using universal probes, rendering this approach reliable for all HA patients carrying a point mutation wherever it is located and chips available for a screening of different HA patients or distinct genes. cSBH is an efficient, reliable, and easy to use re-sequencing technique that offers an alternative approach for screening of large genes. cSBH allows sequencing of large segments of DNA on only one chip, thereby drastically reducing the time it takes to analyze a specific sequence. Actually the mean time to screen the coding regions of FVIII gene was three days. This period was reduced as compared to the time used in standard methods and comparable to that needed for sequencing analysis. The only equipment required, aside from basic tools (e.g., thermal cyclers, pipettes), is access to a standard array scanner, which makes it a very affordable technique to implement. The required data analysis software, which runs on standard PC computers, will be provided as part of the universal HyChip system and is expected to be economically priced. cSBH is a non-radioactive method and generates sequencing results that can be obtained from genomic DNA within a day. Another major advantage of cSBH are the truly universal HyChip arrays which can be applied to screen any DNA sequence without further probe or oligonucleotide changes, which can be very costly. cSBH offers an efficient and accurate screen for most DNA sequences. The cSBH method is highly reproducible because each base is currently read with 22 overlapped probes (11 per strand). In addition, sample preparation and ligation kits, aliquoted for individual assays, are prepared with all components including enzymes to yield very reproducible biochemical reactions. Furthermore, the base-calling software includes several normalization functions to minimize the influence of experimental variations. [13] The combinatorial ligation of universal probe sets coupled with informative probe pools provides a large number of long (currently 11-mers, potentially 12- to 15-mers in two-probe ligation and 17- to 24-mers in three probe ligation), overlapping probes of predetermined behavior that interrogate each base. Robust statistics take advantage of this redundant dataset to provide accurate base calling in spite of array imperfections (0.5-5.0% of missing dots; high droplet size variation), some impaired probe-target hybridizations due to target-target hybridization, significant experimental noise, and the always-present statistical noise related to probe-pooling and sequence repeats. The main software improvement in the future should address target DNA behavior (e.g., primers/primer dimers, palindromes, direct and inverted repeats, and uneven amounts of target DNA) to aid in a more accurate interpretation of base scores and associated P-values. Actually, the presence of short palindromes or repeats may contribute to the number of bases with lower "scores", as seen in sample 2 [Figure 2A]. Furthermore, detection of low-signal AT rich sequences can be improved with optimization of probe concentrations and incorporation of 1,600 AT-rich 7-mers and 8-mers in the arrays, as well as 500 AT-rich 6-mers and 7-mers in the labelled probe pools, which are currently under development. Universal arrays use probes shorter than gene specific arrays. As a consequence, there is a higher chance to have repeated sequences longer than the length of probes (currently 11-mers in cSBH) occurring in a DNA sample. Such sequences may lead to uncertainty in the sequence assembly. For example, a stretch of 12 As can be perceived as 11, 12, 13, or more As. Also, a mutation in the middle of a repeated 20-mer or any longer segment may not be assigned to the actual copy of the repeat. This situation can be avoided by amplicon design that separates repeats into two sequencing reactions. Short identical repeats several bases in length or longer imperfect repeats, both found in exons or immediate intronic sequences that flank each exon, usually do not confuse the advanced base-calling software. There are limitations with cSBH analysis: the current software and probe set must be improved to detect DNA alterations within simple repeat regions, which is a problem common to other sequence technologies as well. [10] Another potential complication, which is common to other high resolution methods, including standard Sanger sequencing, is that all sequence changes, polymorphisms and missense changes that do not result in an obvious pathogenic mutation such as a stop codon and are of unknown clinical significance will be detected. [10] In summary, mutation detection for the FVIII gene requires a multimodal approach that can detect the full range of mutations present in this gene. First-stage mutation screening requires a fast, efficient and possibly economical approach to scan the entire gene. cSBH is a technique that is ideally suited. In this study, we showed that cSBH is capable of detecting a broad range of mutations, currently has the capacity to analyze long continuous read lengths of up to 2 kb per chip, and is able to analyze pooled PCR fragments (such as exons 1-25 in this study) on a single universal chip. References
Copyright 2008 - Indian Journal of Human Genetics The following images related to this document are available:Photo images[hg08013f2.jpg] [hg08013t1.jpg] [hg08013t2.jpg] [hg08013f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}