 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
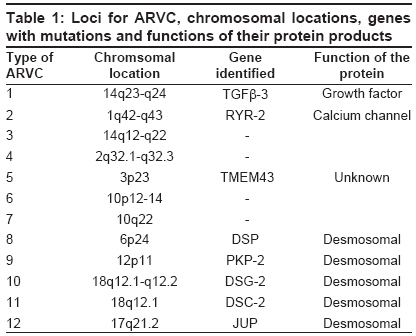
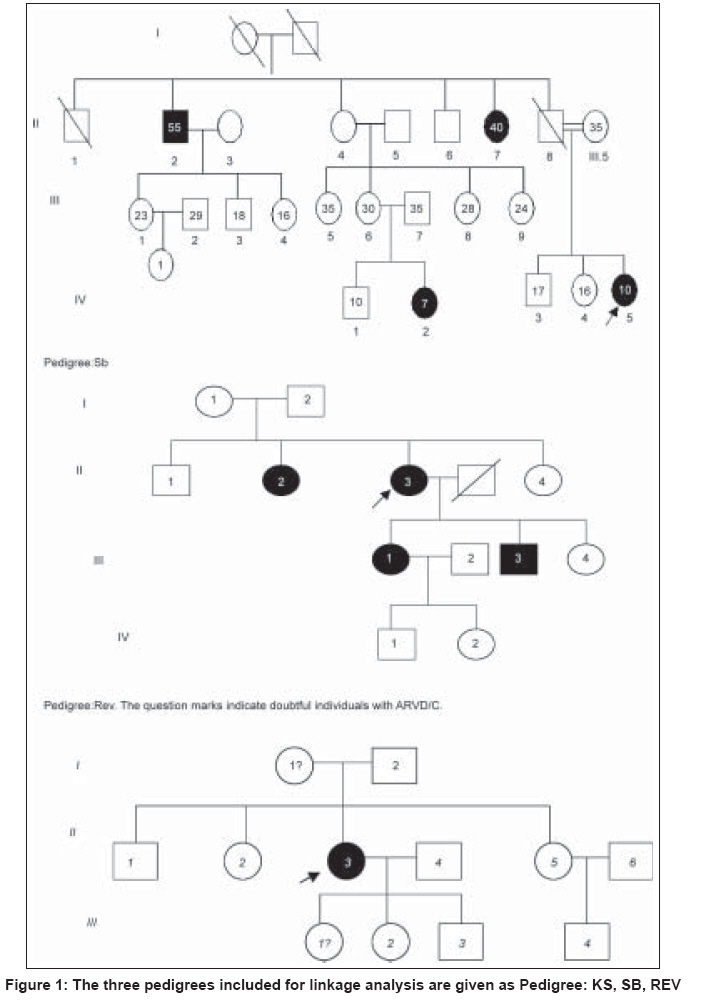
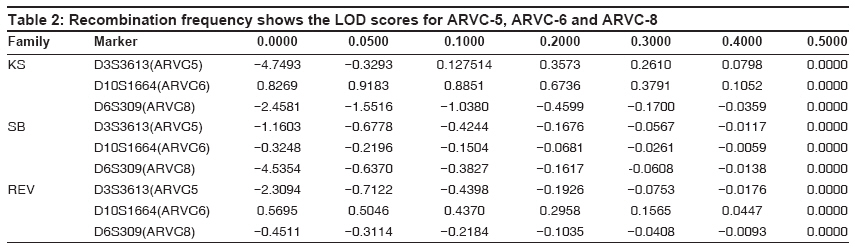
Indian Journal of Human Genetics, Vol. 15, No. 2, May-August, 2009, pp. 54-59 Original Article Linkage analysis of three families with arrythmogenic right ventricular cardiomyopathy in India Dokuparthi MaithiliV. N., Pamuru PranathiR, Oruganti SaiS, Calambur Narsimhan, Nallari Pratibha Department of Genetics, Osmania University, Hyderabad Code Number: hg09014 DOI: 10.4103/0971-6866.55216 Abstract Background: Arrythmogenic Right Ventricular Cardiomyopathy (ARVC) is a primary myocardial disorder morphologically characterized by subtle to severe replacement of the right ventricular myocardium by fatty and fibrous tissue. ARVC is known to be highly prevalent in European population with recent reports implicating it to be a major cause of sudden death in young individuals even from American and Asian population.Aim: To implicate or exclude TMEM43 (ARVC-5), DSP(ARVC-8) genes and the yet to be identified gene at ARVC-6 locus in the pathogenesis in three families affected with ARVC from India. Materials and Methods: Three families comprising of 42 affected/unaffected members were included in the study. Three microsatellite markers, D3S3613 (ARVC5) D10S1664 (ARVC6), D6S309 (ARVC8) were genotyped by PCR-based native PAGE. Two-point Linkage analysis was performed using LINKAGE program version 5.2 . Results and Discussion: LOD scores from linkage analysis for the microsatellite marker D10S1664 (ARVC-6) in families KS and REV have shown positive value hinting the involvement of this locus in the etiology of ARVC, while linkage analysis in the SB family ruled out involvement of DSP, TMEM43 and ARVC-6, as negative LOD scores were obtained with all three loci. Therefore, linkage analysis carried out in the present study indicates that ARVC-6 (cumulative LOD score is equal to plus 1.203376 at q is equal to 0.05) could be the locus harboring the mutated gene in two out of three families. Keywords: Arrythmogenic right ventricular cardiomyopathy, linkage analysis, LOD, sudden death Introduction Arrythmogenic Right Ventricular Cardiomyopathy (ARVC) is a primary myocardial disorder morphologically characterized by subtle to severe replacement of the right ventricular myocardium by fatty and fibrous tissue. The symptoms of this condition may range from palpitations, syncope, pre-syncope to even sudden death as initial manifestation, especially sudden death, without any preliminary symptoms in young adults is the cardinal feature of this condition. To prevent the untimely event of sudden death without any symptoms or signs, early molecular diagnosis is required to proscribe an individual from intense athletic activity which is considered the major trigger of arrhythmic events in many cases. Molecular genetic studies have revealed the association of 12 loci with the condition and among them eight genes have been identified - TGFb-3 (ARVC-1), RYR-2 (ARVC-2), DSP (ARVC-8), PKP2 (ARVC-9), DSG-2 (ARVC-10), DSC-2 (ARVC-11), and JUP (ARVC-12). Recently a gene at ARVC-5 locus called TMEM43 was identified with a misense mutation in all the affected individuals. [1] Details of all the pathogenic loci along with chromosomal locations and genes have been given in [Table - 1]. ARVC is known to be highly prevalent in European populations like Italy where it was first described and reports are emerging from other European, American and Asian populations implicating ARVC to be a major cause of sudden death in young individuals. [2],[3],[4],[5] However, the scenario in the Indian context is obscure. Mutations in the above mentioned genes have been discovered in various populations across the world but the status regarding the genetic etiology in Indian population is unknown. In this study we included individuals belonging to three families and performed Two-point Linkage analysis for ARVC-5, ARVC-6 and ARVC-8 on three families using microsatellite markers at those loci in order to implicate or exclude TMEM43, DSP and the yet to be identified gene at ARVC-6 locus in the pathogenesis in these families. Materials and Methods The present study included three families consisting of 42 affected/unaffected members and these families were approached after the probands were confirmed with ARVC according to standardized diagnostic criteria. [6] Clearance from the Ethics Committee was obtained for the present study and blood samples, clinical and familial information were collected after informed consent from all the participating individuals. The source of these patients was from Care Hospitals, Hyderabad, India. About 5 ml of whole blood was collected from the subjects and DNA isolation was carried out by rapid salting-out DNA isolation protocol. [7] Polymerase Chain Reaction (PCR) was carried out to amplify the three micro-satellite markers (D3S3613, D10S1664, and D6S309) in the vicinity of the loci for ARVC-5, 6 and 8 respectively. Primer sequences were obtained from Genome Databank ( www.gdb.org ). Non-denaturing Polyacrylamide Gel Electrophoresis (PAGE) was used for genotyping the microsatellite markers and the bands were visualized by silver staining. [8] Parametric method of likelihood maximization of classical LOD score analysis was used for linkage analysis. Two-point linkage analysis was performed on a personal computer using LINKAGE program version 5.2. [9] The programs used for two-point linkage analysis were MAKEPED, UNKNOWN and MLINK that were evoked sequentially. The allele frequency of the ARVC locus of normal and mutated sequence was assumed to be 0.9999 and 0.0001 respectively and autosomal dominant mode of inheritance was assumed, since this type of inheritance is well established in isolated form of ARVC. [9] One liability class viz., penetrance was included and the value was set at 95%. [10] Single stranded conformation polymorphism (SSCP) technique was followed for screening 5′-UTRs and 3′UTRs of TGFβ -3, FKBP-12.6 binding domain coding exons of RYR-2 and all the coding exons of PKP-2, DSC-2 and JUP in these three families, to rule out mutations in them. Results Clinical characteristics of families Pedigrees of the three families are given in [Figure - 1] and three families are named as Pedigree.1: KS, Pedigree.2: SB and Pedigree.3: REV. Affected individuals of the families were discovered during the family screening carried out in this study. Pedigree-KS: The proband of the Ped: KS was a 10-year-old girl who had complained of repeated episodes of syncope. The condition was considered extremely serious and heart transplantation was being considered as the next treatment option. However, she expired within a year of diagnosis when she was on medication during the course of this study. When relatives of the proband were enquired about symptoms, most of the members from II- generation reported palpitations and rarely few episodes of syncope. When 12-lead ECG and 2D echocardiography were performed on all the individuals given in pedigree: KS, three members excluding the proband (IV-5) were found to show ECG characteristics indicative of ARVC. These patients showed T-wave inversions with left bundle branch block morphology. Therefore II-2, II-7 (T-wave inversions in V1-V5) and IV-2 (T-wave inversions in anterior pre-cordial leads) satisfied one- major criteria of confirmed case of ARVC in the family and two minor criteria of T-wave inversions in right pre-cordial leads and a sudden death of a relative (father of proband (II-8)) at about 30 years of age without any known cause. Two affected patients from II- generation showed relatively mild symptoms and higher survival age in contrast to two affected individuals of third generation where the diagnostic features were observed severely at less than 10 years of age. Pedigree-SB: The proband (II-3) of Ped: SB was 53-year-old female showing the symptoms of dyspnea on exertion (Grade III) and palpitations (Grade II) with the diagnosis being done two years ago. Magnetic resonance imaging (MRI) revealed abnormalities of right ventricular morphology and 24 hour Holter showed VT (Ventricular tachycardia) from right ventricular outflow tract. The first relative detected to be ARVC patient was the proband′s sister (II-2) who was 54-year-old. She showed no symptoms such as dyspnea, palpitations or syncope but her 2D Echo showed slightly dilated right ventricle with normal systolic function, mild to moderate tricuspid regurgitation. The gross right ventricular morphology showed aneurysms and right atrium also showed enlargement (5.3 x 3.1 cms). Therefore the phenotype of right heart of II-2 is typical of an ARVC patient and comparable to that of II-3 but the patient was asymptomatic. The daughter of the proband (III-1) 26-year-old, upon enquiry for symptoms revealed that she experienced palpitations and syncope from 17 years of age. Her 2D-Echo revealed dilated right ventricle along with dilated right ventricular outflow tract (RVOT) and right atrium. The right ventricular free wall was hypokinetic with local out pouching, a characteristic typical of ARVC. Another member of this family found to be affected during the course of the study was the son of the proband (III-3), who did not experience any symptoms of cardiac abnormality such as dyspnea, palpitations, syncope.etc. His echocardiogram revealed dilated right ventricle with mild TR-grading (tricuspid regurgitation) and slightly dilated right atrium. Therefore this family can be considered a typical pedigree with ARVC with all diagnosed individuals fulfilling the standardized criteria. Pedigree-REV: The proband (II-3) of pedigree: REV was a 39 year-old female with her ECG showing heart rate of 76 beats per minute, normal axis of deviation with sinus rhythm. The QRS complex was found to be prolonged and T-wave inversions were found in the right precordial leads. The relatives of proband, I-1 and III-1 showed minor abnormalities in ECG and 2D-Echo. These were considered as being probably affected in the linkage analysis, as there have been reports for the need to broaden the diagnostic criteria in the affected relatives. [11] Linkage analysis Genotyping of the microsatellite markers has revealed the presence of 10 alleles for ARVC-5 marker D3S3613 and seven alleles for ARVC-6 marker D101664 and seven alleles for marker D6S309 of ARVC-8. The distribution of these alleles in the population was considered to be 1/n (n is equal to number of alleles). Linkage analysis revealed that, linkage with ARVC-5 can be conclusively excluded in the family-KS with LOD of minus 4.749302 and the cumulative LOD of minus 8.219135 (at q is equal to 0.00) indicates that locus for ARVC-5 can be eliminated to be involved in ARVC in these three families. Fam: KS shows slightly positive value plus 0.826904 and Fam: REV plus 0.569591 indicating some kind of possibility of linkage of ARVC-6 in these two families. The cumulative LOD score of plus 1.07161 at q is equal to 0.00 and plus 1.203376 at q is equal to 0.05gives the scope for screening this region further for mutations. Further, linkage analysis with ARVC-8 reveals that DSP can be ruled out to possess mutations in Families-KS and SB with LOD of minus 2.450 and minus 4.500 at q is equal to 0.00 respectively. LOD scores of this analysis have been presented in [Table - 2]. Mutation screening SSCP analysis followed by direct sequencing of samples showing abnormal band pattern, did not show patient specific DNA sequence variants in these families. Reported polymorphisms and intronic variants were observed in probands and controls and none of them were associated with the affected individuals of a family. Discussion Since the familial nature of ARVC is not very well known in the Indian population, we performed the clinical as well as genetic screening of three families who gave consent to perform the study. Clinical evaluation revealed that in Family: KS, the age of onset of the clinical symptoms was decreasing from second generation (onset of symptoms from about 35 years of age) to fourth generation (less than 10 years of age). Further, the proband died during the course of the study even when under medical supervision and IV-2 showed clinical signs in ECG and Echocardiography during the family screening in this study. As this individual was only seven years old, she was not able to answer any questions relating to symptoms. This trend of early age of onset points towards genetic anticipation operating in this family. Another observation in this family was that, though isolated ARVC has been observed to follow only autosomal dominant mode of inheritance, severe expressivity in the form of sudden death in the case of proband (IV-5) at the age of 10 years could be due to consanguinity of parents which may have caused homozygosity of the genetic modifiers. LOD score from linkage analysis in this family (Pedigree: KS) has shown slightly positive value (+0.826904) hinting the involvement of this locus in the etiology of ARVC in this family. In order to rule out mutations in other known genes, 5′-UTR and 3′UTR of TGFβ -3gene in which only regulatory mutations have been reported so far along with FKBP-12.6 binding domain coding exons of RYR-2 and all exons of PKP-2, DSC-2 and JUP were screened in this family. No DNA sequence variants were found in these genes that were specific to affected individuals. Therefore ARVC-6 is a good candidate locus in this family. The second family that we screened (Pedigree:SB) was a typical example for hidden nature of ARVC, as among the 10 members of the family that were clinically screened, four members were detected with the condition upon ECG, Echocardiography and MRI, though none of them considered themselves seriously symptomatic and had no problem in performing all the activities of day to day life. ARVC in this family appeared to be highly penetrant though identification of a genetic lesion would have given a conclusive evidence regarding penetrance. Linkage analysis in this family ruled out involvement of DSP, TMEM43 and ARVC-6, as negative LOD scores were obtained with all three loci. In this family, 5′ and 3′ UTRs of TGF β -3gene, FKBP-12.6 domain coding exons of RYR-2, PKP-2, JUP and DSC-2 were screened and only a reported intronic polymorphism in PKP-2 was observed. As we screened all the coding regions of other genes, the probable mutation could have been in the regulatory regions and the intronic regions that have not been screened in this study. In the third family (Pedigree: REV), both the mother and daughter of the proband showed signs of ARVC, but did not me et al l the diagnostic criteria required for confirmed diagnosis. These were included in the linkage analysis based on the reports of the need for broadening the diagnostic criteria for relatives of the proband as they may show subtle symptoms without any reduction in risk of sudden death. [11] Linkage analysis has shown positive LOD score for ARVC-6 (0.569591) in this family. As mentioned earlier, other genes were screened for mutations, but no patient specific variations could be identified. Therefore, linkage analysis carried out in the present study indicates that ARVC-6 (cumulative LOD score is equal to plus1.203376 at q is equal to 0.05) could be the locus harboring the mutated gene in two out of three families. Among the genes located in the region 10p12-p14, functional candidate gene approach would implicate gene Nebulette (NEBL) at 10p12 that could be involved in the pathogenesis. The cardiac isoform of this protein is known to be involved in the myofibril organization and function and particularly in stabilization of cardiac Z-line. [12] Apart from playing a sarcomeric role, NEBL is also known to have been expressed during early cardiac development playing a role in myofibrillognesis. The isoform of this protein named LIM-Nebulette is known to interact with the versatile component of focal adhesion of eukaryotic cells called Zyxin. [13] The relation of focal adhesion complexes with desmosomal components, which are major pathological sites for ARVC, and the role of Zynix and LIM-Nebulette in cross talk with desmosomal function and regulation would help us understand the role of NEBL in pathogenesis of ARVD/C. Therefore NEBL is a very good candidate that could be harboring mutations in these families. One of the L-type voltage gated Calcium channels called CACNB2, with β -subunit being coded by the gene at 10p12, i.e., at the ARVD/C-6 locus has already been implicated for inherited cardiac arrhythmic syndrome called Brugada syndrome. [14] Therefore this gene could also be a candidate gene for ARVD/C as the function of this gene is homologous to that of RYR-2 (ARVD/C-2). In conclusion, we have excluded the involvement of ARVC-5 and ARVC-8 in the pathogenesis of ARVC in the three families included in the present study, and according to our knowledge this is the first genetic study from India. Acknowledgment This project has been funded by the Council for Scientific and Industrial Research (CSIR) New Delhi. Ms. Maithili.D.V.N is the recipient of CSIR-JRF and SRF. References
Copyright 2009 - Indian Journal of Human Genetics The following images related to this document are available:Photo images[hg09014t2.jpg] [hg09014f1.jpg] [hg09014t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}