 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
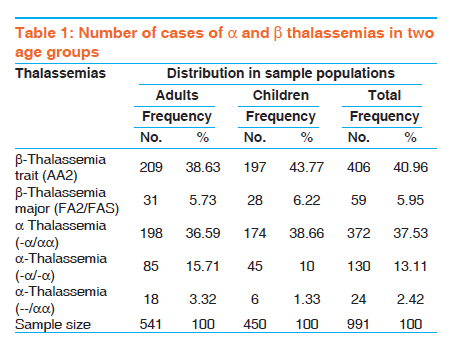
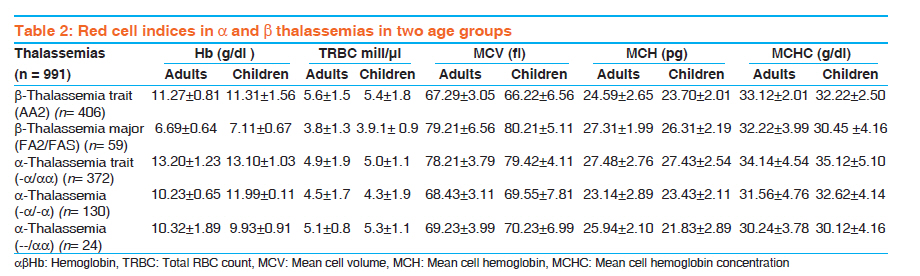
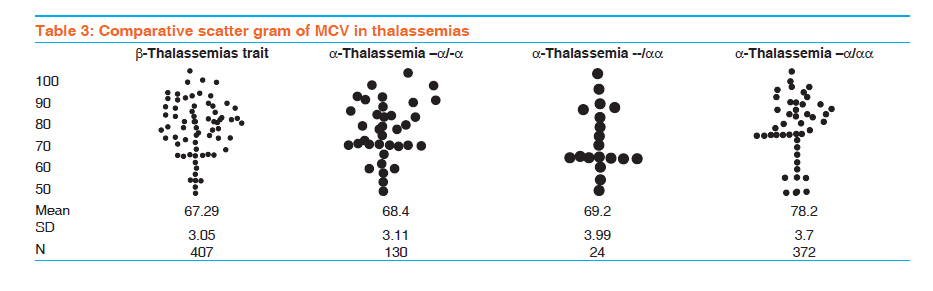
Indian Journal of Human Genetics, Vol. 17, No. 3, September-December, 2011, pp. 207-211 Original Article A comparative study of hematological parameters of α and β thalassemias in a high prevalence zone: Saudi Arabia Syed Riaz Mehdi, Badr Abdullah Al Dahmash Department of Medical Laboratory, Medical College, King Saud University, Riyadh-11416, Saudi Arabia Code Number: hg11043 DOI: 10.4103/0971-6866.92106 Abstract Background and Aims : Saudi Arabia falls in the high prevalent zone of αα and β thalassemias. Early screening for the type of thalassemia is essential for further investigations and management. The study was carried out to differentiate the type of thalassemia based on red cell indices and other hematological parameters. Keywords: Discriminatory index, hematological parameters, Saudi Arabia, thalassemia Introduction Normal hemoglobin A consists of two α and two β chains. The globin gene clusters are present on chromosome 16 while the β gene clusters are present on chromosome 11. Thalassemias are autosomal recessive disorders. In thalassemia, one of the globin chains syntheses may be defective either due to mutation or deletion resulting in excess production of the other chain which damages the red cell membrane. On the basis of the affected globin chain, the thalassemias are classified into α or β types. The β thalassemia is prevalent throughout the world while α is found more in the Mediterranean region, [1] Middle East, [2],[3],[4] South Asia [5],[6] and South East Asia. [7],[8],[9] The hematological parameters in the thalassemic patients vary with the type and severity of anemia. However, varying degrees of microcytosis is invariably a common feature of almost all types of thalassemias. The final diagnosis of the type of thalassemia is made after a battery of laboratory tests like complete blood count, peripheral smear study, hemoglobin electrophoresis, high performance liquid chromatography (HPLC), isoelectric focusing and DNA techniques like PCR, restriction fragment length polymorphism (RFLP). However, the facilities for DNA study are not available at each and every hospital. Some of these tests are cumbersome and expensive. The blood picture and the study of hematological parameters are the key to decide further course of investigation. Many discriminatory indices are available to differentiate β thalassemia trait from iron deficiency anemia (IDA) [10],[11],[12] but no index is described to differentiate clearly between different types of thalassemias itself on the basis of hematological findings. The studies carried out to discriminate between various types of thalassemias on the basis of hematological findings and especially on red cell indices are very limited. [7],[13] The present study is a comparative study of hematological parameters of α and β thalassemias. The study was designed to lay keen emphasis on the differences in the red cell indices of two types of thalassemias. Several studies have been carried out in the past and at different places on thalassemias and their blood picture, [4],[5],[6],[7],[8] but specific comparative studies of hematological parameters done on Saudi thalassemic patients are rare. In Saudi Arabia where some of the regions have a high prevalence of thalassemias, such studies could be significant in early screening of thalassemias. [2],[14],[15],[16],[17] Materials and Methods The present study was carried out in the Hematology department of Riyadh Medical College of Health, Saudi Arabia and its associated hospitals from November 2008 to January 2011. The study sample consisted of 991 cases divided into two groups: adults and children. There were 541 adults and 450 children of both sexes. The blood samples of patients were collected in EDTA vacutainers and run on Coulter STKS hematology analyzer. Complete blood counts including hemoglobin (Hb) and red cell indices; mean cell volume (MCV), mean cell hemoglobin (MCH) and mean cell hemoglobin concentration (MCHC) were estimated. The blood samples were also subjected to cellulose acetate hemoglobin electrophoresis at 8.6 pH. The cases where clear hemoglobin pattern was not observed were reconfirmed by HPLC. Hemoglobin A2 level of >3.5 % was considered as β thalassemia trait. The patients suffering concomitantly with iron deficiency anemia were first treated and then considered for hemoglobin electrophoresis. For gene deletion studies in cases of α thalassemia, RFLP technique using the restriction endonucleases Bam HI was applied. Statistical analysis was carried out by Statistical Package for Social Sciences, (SPSS Inc. Chicago, IL, USA) 11.5 version. For comparison of hematological parameters, Student t-test was used. P value of <0.05 was considered as significant for interpreting the results. Results The average age of the patients in the adult and children groups was 35.6 ± 15.9 and 6.8 ± 3.7 years respectively. The cellulose acetate hemoglobin electrophoresis and gene studies revealed that there were 406 (40.96%) and 59 (5.95%) cases of β thalassemia trait and β thalassemia major respectively including adults and children. The breakup of different gene deletion types of α thalassemias is given in [Table - 1]. The level of hemoglobin was lowest (6.69 ± 0.64) in cases of β thalassemia major. Hetrozygous α-thalassemia trait (-α/αα) showed almost normal levels of hemoglobin and the patients were asymptomatic. Other thalassemia traits like β and homozygous α-thalassemia (--/αα) and (-α/-α) had mild degree of anemia. There was no significant difference in the hemoglobin levels of adults and children in each type of disorder (P < 0.05) [Table - 2]. Moderate degree of microcytosis (MCV = ≤ 78fl) and hypochromia (MCH = ≤ 27pg) was a feature of β thalassemia trait and homozygous α-thalassemias. However, microcytosis was more marked in β thalassemia trait compared to heterozygous α-thalassemias. It was observed that there was statistically no significant difference between the red cell indices of β thalassemia trait and homozygous α-thalassemias (--/αα, -α/-α). (P < 0.01) [Table - 2] [Table - 3] [Table - 4], RBC count was lowest in cases of β thalassemia major and highest in β thalassemia minor. The α-thalassemias showed a normal level of RBC count. β thalassemia major was characteristic of mild degree of microcytosis and hypochromia. Compared to children, the microcytosis was more frequently seen among adults (P <0.023) [Table - 2]. Hetrozygous α-thalassemia trait (-α/αα) was the only disorder in which all the hematological parameters were normal. Here also, there was no significant difference in the findings of adults and children [Table - 2]. Comparative scattergram of values of MCV and MCH in different thalassemias is given in [Table - 3] and [Table - 4]. Discussion Thalassemia is a worldwide disorder. α- and β -thalassemia are the most common single-gene hemoglobin disorders in the world. It is more prevalent in areas endemic for malaria. [18] South East Asia, [7],[8],[9] India, [5],[6] Mediterranean region [2],[3],[4] and Middle East including Saudi Arabia [2],[14],[15],[16],[17] are the regions from where large number of cases are reported. The change in hematological parameters depends on the type of thalassemia. Unlike β -thalassemia trait and iron deficiency, no simple biochemical test can detect α-thalassemia. The key to successful detection and characterization of the hemoglobinopathies, particularly the thalassemias, is the initial hematological data. The clue for thalassemia is low mean corpuscular volume (MCV) < 78 fl or low mean corpuscular hemoglobin (MCH) <27 pg. Although iron deficiency is the most common cause of a low MCV or a low MCH, it is likely that this finding will point to thalassemia in regions of countries with thalassemia-prone ethnic populations.There are several causes of the anemia produced by different abnormal hemoglobins. Microcytic hypochromic anemia is a common hematological abnormality in clinical practice and usually is caused by iron deficiency and thalassemia trait. Red cell indices are taken to be the differentiating factor in anemias due to thalassemias and those from iron deficiency and are very necessary as both occur in the same areas. Relative increase in red cell count in thalassemias gives reduced MCH and MCV values. The degree of microcytosis and type of mutation in thalassemias have shown wide variations in ranges of MCV. [1],[6],[7],[19] There is limited data reported in the literature for prevalence of α-gene deletion in patients with microcytosis. [20],[21],[22] Carriers of α-gene deletion have mild microcytosis with or without anemia. Although anemia is absent or unremarkable, it is important to diagnose α -thalassemia to ascertain the cause of microcytosis and to avoid repeated expensive analysis and/or prolonged iron therapy. The coexistence of α deletions in β -thalassemia patients modifies the phenotype. So far, no biochemical diagnostic test is available for detection of α-thalassemia carriers and globin chain synthesis studies are time consuming.Molecular methods like southern blot hybridization and sequencing are conventionally used for diagnosis of α-thalassemia. However, molecular diagnosis by PCR has proven to be less time-consuming and less expensive, with a more definite outcome in the clinical setup. The most widely used method in detection of β -thalassemia′ mutation is the amplification refractory mutation system (ARMS). Unlike the β -thalassemias, deletions are a common cause of α-thalassemia. The common α-thalassemia deletions and rearrangements can be routinely detected using gap-PCR. [23],[24] Hematological parameters in patients with α -thalassemia were compared with those in patients with β -thalassemia. Individuals with the single-gene deletion had lower levels of hemoglobin, MCV and MCH as compared to normal controls. The carriers of α -thalassemia have a mild microcytic hypochromic anemia as observed by other authors. However, their MCV and MCH were better than those of patients with iron deficiency anemia. [18] MCH is a better discriminator than other red cell indices in the diagnosis of α-thalassemia. [1],[7],[18] Since there is no definitive hematological marker that can give the diagnosis of α -thalassemia, molecular analysis remains the only diagnostic approach in microcytic hypochromic anemia patients. Our findings are in accordance with previous reports, where microcytosis was explained on the basis of number of α-gene deletions. [20],[22] The identification of α and β -thalassemia carrier status is important before going for expensive investigations to define the etiology of anemia, as well as to prevent unnecessary prolonged iron supplementation. Thus screening for thalassemia should be considered during genetic counseling of couples at high risk of thalassemia, for prenatal and premarital diagnosis.Identification of rare thalassemia gene mutations in our population was necessary because of consanguineous marriages in Saudi Arabia. The use of more comprehensive diagnostic tools is more useful in the diagnosis of unresolved cases. However, hematological parameters remain the earliest and most cost effective method in differentiating β thalassemia from Iron deficiency anemia and many discriminating indices are also available to differentiate between the two, but none to differentiate between various types of thalassemias. Conclusion We observed that hematological parameters are helpful in differentiating β thalassemia major from β thalassemias minor but not of much help in differentiating α thalassemias from β. Varying degree of microcytosis is a common feature in both types of thalassemias. MCH is a better indicator in discriminating various α thalassemias. There is a dire need to develop a discrimination index to differentiate α thalassemia from β thalassemias on the pattern of indices available for discriminating β thalassemia trait from iron deficiency anemia. If such an index is developed, it would help physicians to adapt the right protocol to investigate the patients of thalassemias and would reduce the time and cost of reaching the final diagnosis. References
Copyright 2011 - Indian Journal of Human Genetics The following images related to this document are available:Photo images[hg11043t4.jpg] [hg11043t1.jpg] [hg11043t3.jpg] [hg11043t2.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}