 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
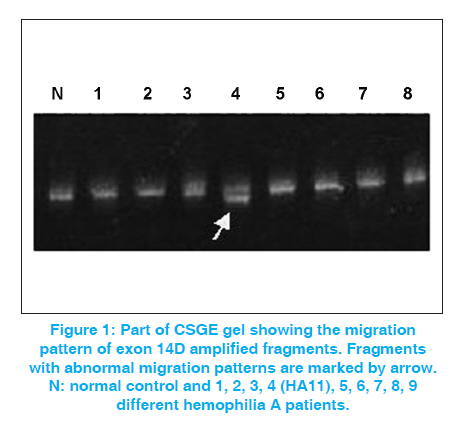
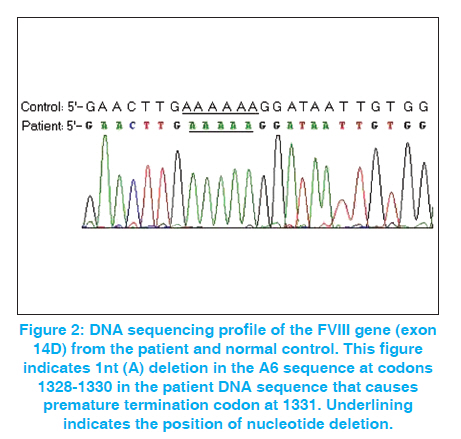
Indian Journal of Human Genetics, Vol. 17, No. 3, September-December, 2011, pp. 232-234 Case Report A novel mutation (4040-4045 nt. delA) in exon 14 of the factor VIII gene causing severe hemophilia A Habib Onsori1, Mohammad Ali Hosseinpour Feizi2, Abbas Ali Hosseinpour Feizi3 1 Department of Biology, Marand Branch, Islamic Azad University, Marand, Iran Code Number: hg11048 DOI: 10.4103/0971-6866.92095 Abstract Hemophilia A is an X-linked congenital bleeding disorder caused by Factor VIII deficiency. Different mutations including point mutations, deletions, insertions and inversions have been reported in the FVIII gene, which cause hemophilia A. In the current study, with the use of conformational sensitive gel electrophoresis (CSGE) analysis, we report a novel 1-nt deletion in the A6 sequence at codons 1328-1330 (4040-4045 nt delA) occurring in exon 14 of the FVIII gene in a seven-year-old Iranian boy with severe hemophilia A. This mutation that causes frameshift and premature stop-codon at 1331 has not previously been reported in the F8 Hemophilia A Mutation, Structure, Test and Resource Site (HAMSTeRS) database. Keywords: Conformational sensitive gel electrophoresis, deletion, frameshift, factor VIII, Hemophilia A, novel mutation Introduction Hemophilia A is an inherited deficiency of coagulation factor VIII (FVIII). It is caused by mutations in the factor VIII gene and its incidence is estimated to be 1:5,000-10,000 in men. [1],[2] Deleterious mutations in the FVIII gene have been demonstrated to reduce either or both activity and circulating plasma level of FVIII protein and thus cause disease. [3] Point mutations are the most prevalent type of defect, probably underlying the disease in 90-95% of patients. Deletions are the second most common gene defects (5-10% of patients). [4] The identification of carriers of the disease is an essential part of genetic counseling and prenatal diagnosis. [5] Case Report A seven-year-old boy (HA11) was referred to our hospital in 2008 due to gum bleeding. Severe bleeding with other medical inspections showed probability of severe hemophilia A. In order to assess the causal mutation, genomic DNA was extracted from 2.5 mL of EDTA anticoagulated peripheral blood by SDS-proteinase K according to Sambrook et al. [6] Polymerase chain reaction (PCR) of genomic DNA from the patient and a normal male subject for all 26 exons with exon-intron boundaries regions were performed in 37 segments with the use of same sets of primers as reported by Steve Keeney. [7] PCR reactions were carried out in 25 μL reaction mixture containing 1 × PCR buffer, 1.5 mM MgCl 2 , 0.2 mM dNTP, 10 pmoles of each primer, 0.5 U of Taq DNA polymerase and about 1μg of genomic DNA on a Genius Thermal Cycler. After initial denaturation for 2 min at 95°C, 30 cycles of 30 s at 94°C, annealing for 40 s at 59°C and extension for 30 s at 72°C, a final extension for 5 min at 72 °C followed. The amplified fragments were detected on 1.5% agarose gel by ethidium bromide staining. The PCR-amplified fragments longer than 400bp were subjected to conformational sensitive gel electrophoresis (CSGE) and the fragments shorter than 400bp were subjected to single-stranded conformational polymorphism (SSCP) analysis. CSGE was performed on 10% acrylamide gel with 1,4-bis-acrolyl piperazine (99:1), 15% formamide and 10% ethylene glycol in 0.5× TTE buffer (1x TTE = 89 mmol/L tris, 28.5 mmol/L Taurin and 0.2 mmol/L EDTA). Ammonium persulfate and N,N,N΄,N΄- tetramethylethylene diamine (TEMED) were used to catalyze polymerization. [8] Samples (heteroduplexes) were prepared by mixing 2 μl of the PCR product from a control DNA with 2 μl of the PCR product of the corresponding fragment, and then incubated at 98°C for 2 min, followed by 10 min at 65°C and 20 min at 37°C. After this step, 5 μl of 10x loading buffer (30% glycerol, 0.25% xylene cyanol, and 0.25% bromophenol blue in distilled water) was added to each sample, which was then applied to the gel. Prior to loading, the gel was electrophoresed at 700 V for 0.5-1h. After loading 5 μl of heteroduplexed amplicons, electrophoresis was carried out at 300 V for 16 h. The gel was stained with ethidium bromide for 15 min. Detection was carried out by UV doc apparatus. SSCP was performed as described previously. [9] PCR products showing abnormal electrophoretic mobility on SSCP or CSGE gel were sequenced by Kawsar Company and analyzed by sequencing-analysis Chromas Lite 2.01 software. The sequences were compared with the wild type. Results In this study, we detected a novel single nucleotide deletion occurring in exon 14 of the FVIII gene in a seven-year-old Iranian boy with severe hemophilia A (FVIII: C <1%). Among all amplified fragments, only PCR product of exon 14D with 595bp long displayed unmatched shift in CSGE [Figure - 1]. Sequence analysis of exon 14D PCR product showed a novel mutation [Figure - 2]. This mutation due to 1-nt deletion in the A6 sequence at codons 1328-1330 (4040-4045 nt delA) causes frameshift and premature stop- codon (termination at 1331). This new mutation was recorded in GenBank (NCBI) with accession number EU 598148.1. Discussion Small deletions and insertions in the coding region of the gene, in most cases, resulting in frameshifts, and more than half of them are in the large exon 14. These mutations with only few exceptions are leading to a severe clinical phenotype. [10] According to the hemophilia A database (http://europium.csc.mrc.ac.uk), from identified 175 small deletion (<50bp) in exon 14 of the factor VIII gene, 91 cases (52%) are 1A deletion that cause severe hemophilia A in all of them. Due to the nature of the sequence alteration, these mutations are expected to be easily detected by different screening approaches with a high rate of sensitivity. [10] In this study, we used CSGE method for mutation detection. It has the advantages of being simple and relatively rapid to perform and does not require the use of radiolabel. Despite this apparent simplicity, the technique requires a great deal of skill, both technical and interpretive, to achieve good sensitivity. So, this study shows that use of CSGE and direct sequencing can be useful for point mutation recognition of the FVIII gene. Acknowledgements This study was supported by Islamic Azad University. The authors thank Dr. Somayeh Akrami and Mehdi Haggi for laboratory cooperation. References
Copyright 2011 - Indian Journal of Human Genetics The following images related to this document are available:Photo images[hg11048f2.jpg] [hg11048f1.jpg] |
| |||||||||

{kind=link}
{kind=link}