 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Human Genetics, Vol. 18, No. 1, January-April, 2012, pp. 134-136 A homozygous female hemophilia A Preethi S Nair, S Shetty, Kanjaksha Ghosh Department of Thrombosis Haemostasis, National Institute of Immunohaematology, KEM Hospital Campus, Parel, Mumbai, Maharashtra, India Code Number: hg12028 DOI: 10.4103/0971-6866.96685 Abstract Background: Hemophilia A (HA), being an X-linked recessive disorder, females are rarely affected, although they can be carriers.
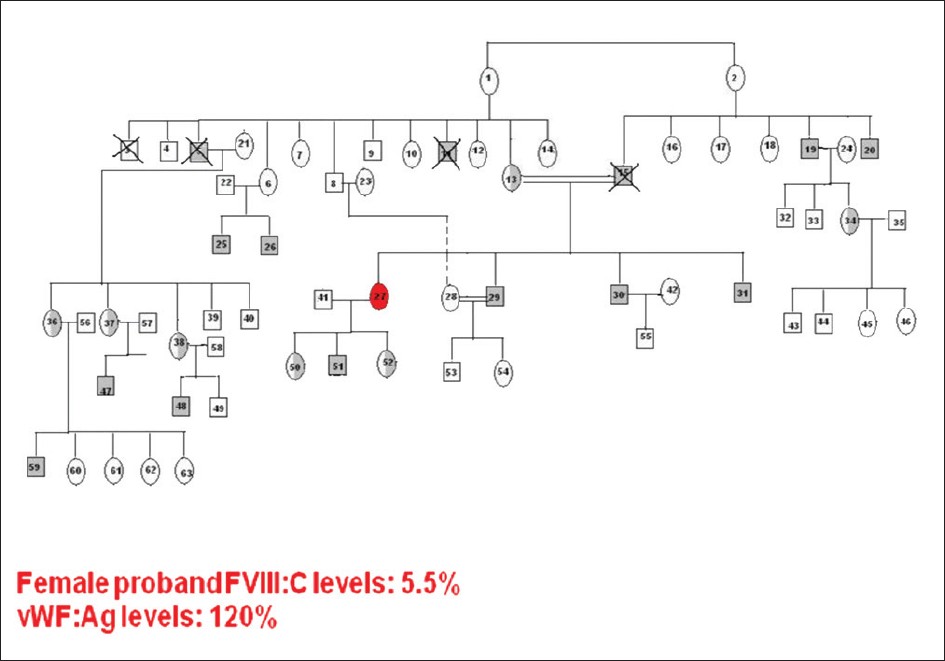
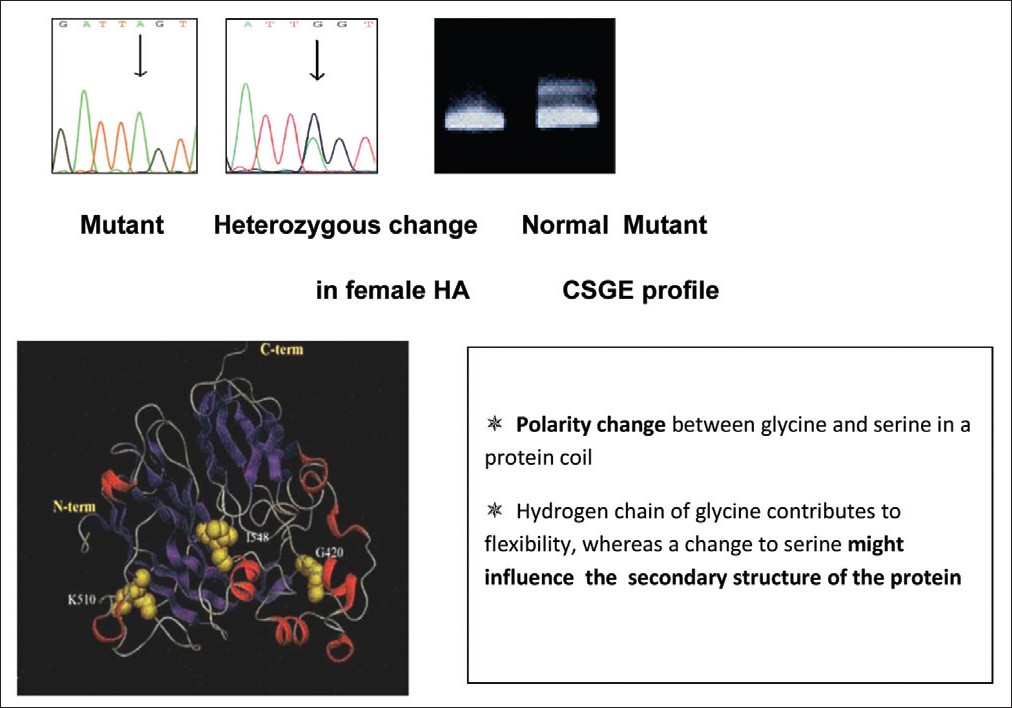
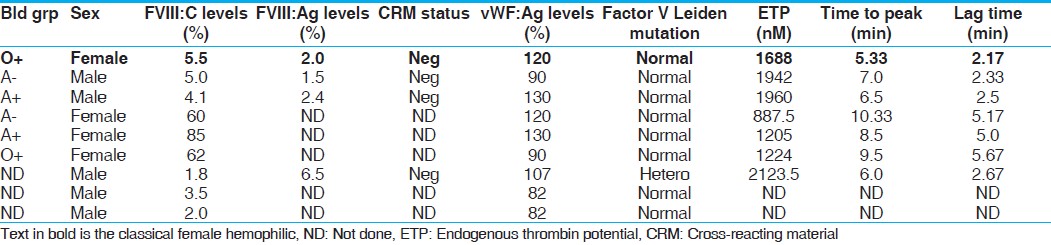
Keywords: Consanguinity, factor VIII, female hemophilia, India Introduction Hemophilia A (HA) results from the partial or complete deficiency of functional factor VIII (FVIII) protein caused by a wide range of heterogeneous mutations in the factor 8 (F8) gene. [1] Being an X-linked recessive disorder, females are generally not affected, although they can be carriers of this disorder. A classical female hemophilia is possible only when a carrier female marries a hemophilic male, of which there are only few reports in the literature. [2],[3],[4] Few other reports of homozygous female HA are mainly due to de novo mutations in one or both the X chromosomes. [5],[6]Materials and Methods Blood was collected by peripheral venous puncture at a 1:10 volume ratio in 3.8% trisodium citrate. Activated partial thromboplastin time (aPTT) was determined according to standard techniques. Factor VIII activity (FVIII:C) of each sample was determined by one-stage aPTT-based assay in a semiautomated coagulometer (Diagnostica Stago, Asnieres, France). FVIII inhibitor activity was determined using Bethesda assay. Plasma von Willebrand factor antigen level (VWF:Ag) was measured by enzyme-linked immunosorbent assay (Diagnostica Stago). DNA was extracted using the phenol-chloroform extraction method. Multiplex polymerase chain reactions spanning all the 26 exons were carried out [7] and conformation-sensitive gel electrophoresis (CSGE) [8] was used to screen the mutations. Amplicons were run on a 12.5% gel (acrylamide: 1, 4-bis acroylyl piperazine - 99:1) incorporating mild denaturants (10%) ethylene glycol, 15% formamide and electrophoresed overnight at 80V. Samples with altered mobility were subjected to DNA sequencing to confirm the mutation. DNA sequencing was performed on both strands using the Big Dye Terminator Cycle Sequencing V1.1 Ready Reaction kit and an automated ABI DNA sequencer (Applied Biosystems, Carlsbad, California. U.S.A).Results We report a case of homozygous female HA with moderate FVIII deficiency (moderate FVIII deficiency 5.5% FVIII:C, vWF:Ag 120%), born of a 1° consanguineous marriage. She presented to our center with severe bleeding diathesis following delivery of her first child and a history of prolonged bleed from cuts and injuries. The mother (a carrier of HA) of the proband was married to a hemophilic who died of excessive bleeding in a road accident and was undiagnosed but his two brothers were diagnosed with moderate deficiency of HA (F VIII:C 4.8% and 5.7%). Her two brothers were affected and the sister was found to be a carrier of HA.[Figure - 1] shows the family pedigree of this patient. Sequencing of this exon revealed a missense mutation corresponding to c.1315G>A, which results in the amino acid substitution corresponding to p.Gly420Ser that fits well with the clinical manifestation of the patient [Figure - 2]. The mother was shown to be a heterozygous carrier of the mutation, while the two affected brothers, uncles and nephews also showed the same mutation. They were found to be cross-reacting material (CRM) negative [Table - 1]. Discussion It was inferred that the father of this index case was also affected and carried the same mutation as that of the index case. The mutation has earlier been reported to be associated with moderate and severe HA, causing polarity change in the protein coil thus influencing the secondary structure of the protein. [9]Female carriers of HA normally do not exhibit a moderate to severe phenotypic expression of the disease. However, a number of other pathophysiological mechanisms may account for the phenotypic expression of very low FVIII:C levels in females. These include (i) skewed inactivation of the X chromosome leading to predominant expression of the mutated allele as a result of a preferential inactivation of the X chromosome with the wild-type F8 gene, [10] (ii) Turner syndrome, (iii) translocation or (iv) males with a female phenotype due to mutation in the sex-determining region Y (SRY) gene on the Y chromosome, i.e. Swyer syndrome combined with a mutation in the F8 gene. In the present report, we have presented homozygosity of the F8 gene mutation resulting in clinical manifestation of HA in a female. In India, as consanguineous marriages are very common in certain communities (up to 30%), the likelihood of encountering female hemophilia is higher, although this is the first case of HA encountered out of 1600 hemophilia families registered in our Comprehensive Haemophilia Care center. Genetic diagnosis in such cases is not necessary as all the male children will be affected and daughters are obligatory carriers. Acknowledgment The authors wish to express their gratitude for The Lady Tata Senior Fellowship, granted by The Lady Tata Memorial Trust, Mumbai, to one of the authors (P. Nair) and the DST funding agency for approving and funding this study.References
Copyright 2012 - Indian Journal of Human Genetics The following images related to this document are available:Photo images[hg12028f2.jpg] [hg12028t1.jpg] [hg12028f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}