 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
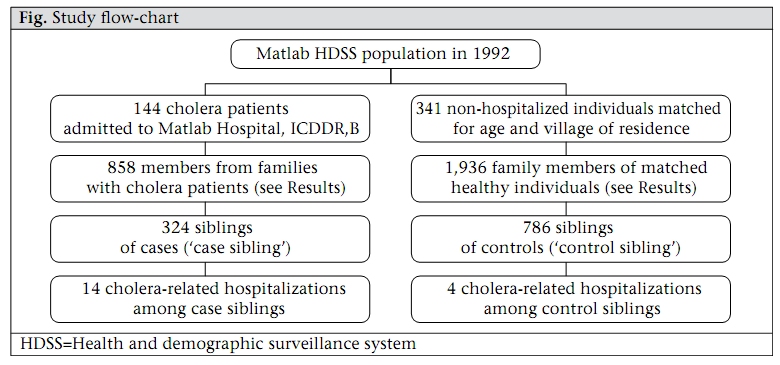
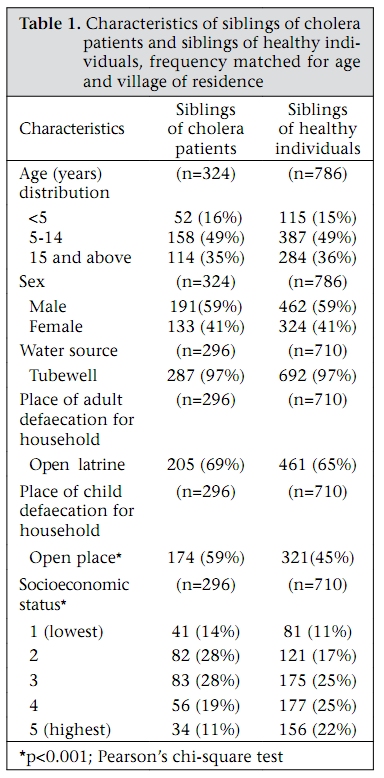
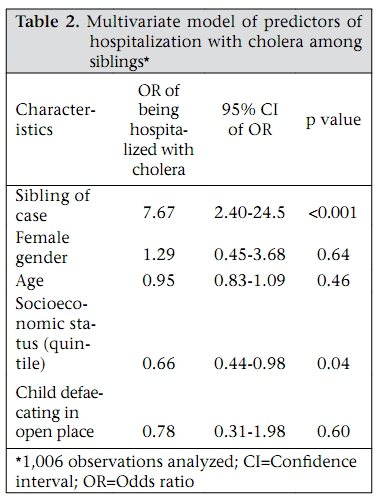
Journal of Health, Population and Nutrition, Vol. 27, No. 6, Dec, 2009, pp. 725-732 Familial Aggregation of Vibrio cholerae-associated Infection in Matlab, Bangladesh Kazi Mizanur Rahman1, Priya Duggal2, Jason B. Harris3, Sajal Kumar Saha1, Peter Kim Streatfield1, Edward T. Ryan3, Stephen B. Calderwood3, Firdausi Qadri1, Mohammad Yunus1, and Regina C. LaRocque3 1ICDDR,B, GPO Box 128, Dhaka 1000, Bangladesh, 2National Human Genome Research Institute, National Institutes of Health, Baltimore, MD, USA, and 3Division of Infectious Diseases, Massachusetts General Hospital, Boston, Massachusetts, USA Correspondence and reprint requests should be addressed to: Dr. Kazi Mizanur Rahman, Child Health Unit, Public Health Sciences Division, ICDDR,B, GPO Box 128, Dhaka 1000, Bangladesh Email: mizan@icddrb.org Fax: 880-2-8826050 Code Number: hn09075 ABSTRACT Vibrio cholerae is a major cause of diarrhoeal illness in endemic regions, such as Bangladesh. Understanding the factors that determine an individual’s susceptibility to infection due to V. cholerae may lead to improved prevention and control strategies. Increasing evidence suggests that human genetic factors affect the severity of V. cholerae-associated infection. This study, therefore, sought to characterize the heritable component of susceptibility to infection due to V. cholerae using the Matlab Health and Demographic Surveillance System database of the International Centre for Diarrhoeal Disease Research, Bangladesh. In total, 144 pedigrees that included a cholera patient and 341 pedigrees without a cholera patient were evaluated during 1 January–31 December 1992. The odds of the sibling of a patient being admitted with cholera were 7.67 times the odds of the sibling of an unaffected individual being admitted with cholera [95% confidence interval (CI) 2.40-24.5, p<0.001], after adjustment for gender, age, socioeconomic status, and hygiene practices. Although exposure to environmental reservoirs is essential in the epidemiology of cholera, household-specific factors, such as familial relatedness to an index case, may also be important determinants of risk of cholera. Further analysis of human genetic factors that contribute to susceptibility to cholera may be productive. Key words: Case-control studies; Cholera; Familial aggregation; Risk factors; Vibrio cholerae; Bangladesh INTRODUCTION Vibrio cholerae, a Gram-negative bacillus, causes a severe dehydrating diarrhoea that may be fatal if timely treatment is not provided (1). Much of the impact of cholera is in India and Bangladesh where cholera exists in both epidemic and endemic forms. Explosive epidemics of cholera have also occurred in refugee-camp settings in sub-Saharan Africa (2-4), and an ongoing epidemic is occurring in Zimbabwe (5). A greater understanding of the factors that determine susceptibility to infection due to V. cholerae, particularly in endemic settings, is needed. Environmental factors, such as plankton blooms, temperature oscillations, and vibriophage dynamics, appear to play a role in inciting epidemics of cholera and have been the subject of a number of recent reviews (6-8). Host factors, such as nutritional status and adaptive and innate immune responses, are also important determinants but remain incompletely understood (9-11). Mounting observational and experimental evidence suggests that human genetic factors affect the severity of V. cholerae-associated infection. In a study of 410 patients from Matlab, Glass and colleagues identified a significant association between blood group O and severe cholera infection (12). This finding was reproduced in a study of V. cholerae-associated infection performed in North American volunteers where blood group O was found in 64% of volunteers with severe cholera versus 36% of volunteers with mild or no illness (13). Interestingly, the lowest prevalence of blood group O in the world was found among persons living near the Ganges River delta where cholera is endemic (14). The mechanism of the observed association between the blood group O and the susceptibility to severe cholera infection remains unclear. Most recently, a family-based candidate gene-association study in Bangladesh identified an association between cholera and a variant in LPLUNC1, an innate immunity protein (15). The sequencing of the human genome has made it possible to identify additional human genetic polymorphisms associated with infectious diseases (16). Such associations can shed light on the role of host factors in the pathogenesis of infectious diseases and may lead to the development of tests for disease susceptibility and improved drug treatments or vaccines. Further studies are needed to understand the role of human genetics in susceptibility to infection due to V. cholerae. As a prelude to such work, we sought evidence of a heritable component of susceptibility to severe cholera infection among households participating in the Health and Demographic Surveillance System (HDSS) in Matlab, Bangladesh. MATERIALS AND METHODS Since 1966, the International Centre for Diarrhoeal Disease Research, Bangladesh (ICDDR,B) has maintained the HDSS, including approximately 220,000 individuals living in Matlab, located 60 kilometres southeast of Dhaka. Health and demographic data are collected systematically through regular household-visits. Hospital-level care for individuals in the HDSS population with diarrhoeal illness is provided by the ICDDR,B hospital in Matlab; records of hospitalizations at the Matlab Hospital are linked to the HDSS dataset via a unique identifier. We used a family case-control study design to assess the familial aggregation of cholera among participants in the Matlab HDSS. In particular, we focused on the risk of V. cholerae-associated infection among siblings of hospitalized cholera patients compared to the risk among siblings of healthy individuals. We chose to evaluate the risk among siblings, rather than among parents because siblings are closer in age and more likely to have similar levels of existing immunity to V. cholerae. During 1 January–31 December 1992, 721 individuals with cholera from the Matlab HDSS population were admitted to the Matlab Hospital. All the cholera patients were confirmed to have infection due to V. cholerae O1 El Tor by culturing a stool specimen or rectal swab at the laboratory of the same hospital. For this study, we randomly selected 300 of these hospitalized individuals, none of whom had more than one hospitalization with cholera during 1992. We selected the year 1992 because it was an epidemic year before the emergence of the new epidemic serogroup—V. cholerae O139. For each hospitalized patient, 2-3 healthy individuals who were not admitted with cholera during that period and who were frequency-matched to the hospitalized patient for village of residence and for age within three years were identified. For the familial aggregation study, cases (‘case siblings’) were defined as siblings of a hospitalized cholera patient, and controls (‘control siblings’) were defined as siblings of a frequency-matched healthy individual. These siblings were identified through the HDSS database; pedigree data could only be constructed for 144 of the selected hospitalized patients and 341 of the healthy individuals. Control siblings were included only once in the study. Information from the Matlab HDSS regarding the source of water and hygiene habits for each case sibling and control sibling was collected. All family members shared the same water-source and sanitary facilities. Data on socioeconomic status were obtained from a survey of the Matlab population performed in 1996, in which households were categorized into five economic strata based on type of household possessions and land ownership. Approval for the present research was obtained from the Research Review Committee of the ICDDR,B and the Institutional Review Board of the Massachusetts General Hospital. All analyses were performed on data lacking any personal identifiers. Demographic characteristics of the case and control siblings were compared using the Pearson’s chi-square test. For multivariate analysis, we used the generalized estimating equations (GEEs) (Intercooled Stata, version 9.2), clustered by pedigree with an exchangeable correlation matrix. A GEE analysis accounts for the dependence of observations within families (17-19). Marginal odds ratios (ORs) and 95% confidence intervals (CIs) were computed for each logistic regression model using a robust method. Missing values for socioeconomic status and defaecation practices of children were dropped from the multivariate GEE analysis, assuming that these were missing at random. RESULTS One hundred forty-four pedigrees that included a cholera patient and 341 pedigrees that included a healthy individual, frequency matched to the cholera patient for age and village of residence, were used in the analysis. As outlined in Figure 1, 858 individuals were members of families with cholera patients, and 1,936 individuals were members of families with healthy individuals. Among these family members, 324 case siblings and 786 control siblings were identified; risk factors for cholera were assessed in this group of siblings. Table 1 shows the characteristics of the case and control siblings. Age distribution was similar in the two groups, and almost half of the individuals were aged 5-14 years. There were more males than females in both the groups. The case and control siblings were similar in terms of water-source and sanitation practices. Tubewell water was consumed by 97% of individuals. The majority of adults in both the groups used open latrines (Table 1). Among children, defaecation in an open place was more commonly practised in the case households compared to the control households (p<0.001). The proportion of individuals in the most advantaged socioeconomic category was higher among the control siblings than among the case siblings (p<0.001). As outlined in Figure 1, 14 case siblings were admitted to the Matlab Hospital with culture-confirmed cholera during 1 January–31 December 1992, and four control siblings were hospitalized during the same period. A median of three days (range 0-9 days) separated the dates of hospitalization of the index patient and his or her sibling. On univariate GEE analysis, the odds of a case sibling being admitted with cholera were 8.83 times the odds of a control sibling being admitted with cholera (95% CI 2.89-27.00, p<0.001). There was a slight reduction in this OR when gender, age, socioeconomic status of the sibling, and open-space defaecation by children in the household were included in a mul-tivariate model (Table 2), but the association remained significant. Of note, lower socioeconomic status remained a significant predictor of hospitalization with cholera in multivariate analysis, although the OR was less than that of being a case sibling. DISCUSSION V. cholerae is an aquatic organism, and exposure to environmental reservoirs is considered important in the epidemiology of cholera. Here, we have demonstrated that household-specific factors—particularly socioeconomic status and being a sibling of an index case—are important determinants of risk of cholera. The mechanism of these associations could not be established in the present study but our results raise hypotheses that merit further research. It is well-established that Vibrio species survive in the aquatic environment in association with zooplankton and phytoplankton and that seasonal increases in the incidence of cholera occur in endemic areas, perhaps in association with a bloom of these natural reservoirs (20). We attempted to account for this by selecting control households from the same geographic area and period as case households, and hence, factors relating to the aquatic environment are less likely to have accounted for our findings. Contamination of household drinking-water or person-to-person transmission of the organism may have accounted for the familial clustering of cholera observed in our study population. Virtually, all study individuals obtained drinking-water from tubewells which are considered much less likely to contain V. cholerae than surface-water sources. Nevertheless, it is possible that tubewell water may have been contaminated as a result of adjacent defaecation practices; our finding of more open-place defaecation among children in the case households supports this possibility. Alternately, the quality of drinking-water may have been compromised by hygiene practices within the household. Direct spread of V. cholerae may also have occurred between family members within a household. Recent mathematical modelling data suggest that person-to-person transmission of the organism, including that from asymptomatic individuals, may play a more important role in the dynamics of outbreaks of cholera than previously appreciated (21). In the present study, a higher socioeconomic status predicted a lower risk of hospitalization with cholera among siblings, and this may reflect better hand-hygiene or better practices in the handling of drinking-water within the home. Further studies aimed at characterizing household-associated practices that facilitate the transmission of V. cholerae in Matlab are worthwhile and may identify potential areas for intervention. The strongest predictor of hospitalization with cholera in our study population was being a sibling of a case patient with cholera, and this association persisted after adjustment for age, gender, defaecation practices, and socioeconomic status. This aggregation of cholera within siblings suggests that shared genetic factors may play a role in susceptibility to V. cholerae. A number of epidemiologic studies have established that genetic variation in the human population contributes to susceptibility to infectious diseases, such as malaria, tuberculosis, leprosy, and leishmaniasis (22). Less attention has been paid to the role of human genetics in susceptibility to diarrhoeal diseases such as cholera, although observational evidence suggests that factors, such as blood group and the innate immune system, may play a role. Although not strictly a genealogical database, the Matlab HDSS proved a useful tool here for establishing familial aggregation of cholera. Our results suggest that further studies, using techniques based on the sequencing of the human genome, are worthwhile to further elucidate the role of host genetics in susceptibility to cholera. A few limitations of our study deserve mention. Our analysis focused only on patients who were admitted to the Matlab Hospital of ICDDR,B with culture-positive V. cholerae-associated infection; individuals with less severe cholera who did not require care at the Matlab Hospital were not represented in our analysis. Pedigree structure could not be ascertained for all selected families, and socioeconomic data were collected outside the study period. Any of these factors may have introduced unrecognized biases into our results. Since our study was a secondary analysis of an existing database, we did not have potentially-relevant information regarding the population’s pre-existing immunity to V. cholerae, history of cholera infection, or blood group phenotype. We did, however, adjust for numerous factors known to be associated with infection due to V. cholerae in our multivariate analysis. The results of our analysis of V. cholerae O1 may not be generalizable to the more recently-emerged O139 serogroup (23); a familial aggregation study focused on this pathogen would be of interest. In conclusion, we used a population-based demographic database to demonstrate the presence of familialaggregationofcholerainMatlab,Bangladesh. Our results confirm the results of a recent study that identified an increased risk of cholera among first-degree relatives living in the same household of an index patient compared to more distantly-related individuals (11). Although this clustering of cholera among siblings may be related to shared environmental or behavioural factors, our results suggest that further analysis of the role of human genetic variability in susceptibility to cholera may be productive and that family-based designs would be appropriate for such work. ACKNOWLEDGEMENTS This research was funded by the Fogarty International Center of the National Institutes of Health (NIH) (Grant No. D43-TW05572 for K.M.R.), the Intramural Program of the National Human Genome Research Institute of the NIH (P.D.), the Fogarty International Center of the NIH (Grant No. K01-TW07409 for J.B.H.), the National Institute of Allergy and Infectious Diseases (Grant No. U01-AI58935 for S.B.C.), the National Institute of Allergy and Infectious Diseases (Grant No. R03-AI063079 for F.Q.), a Claflin Distinguished Scholar Award from the Massachusetts General Hospital (R.C.L.), and the Fogarty International Center of the NIH (Grant No. K01-TW07144 for R.C.L.). ICDDR,B acknowledges with gratitude the commitment of the above-mentioned institutes to the Centre’s research efforts. REFERENCES
Copyright 2009 - International Centre For Diarrhoeal Disease Research, Bangladesh The following images related to this document are available:Photo images[hn09075t1.jpg] [hn09075f1.jpg] [hn09075t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}