 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Health Population and Nutrition, Vol. 28, No. 6, December, 2010, pp. 545-552 Quantifying the Association between CampylobacterInfection and Guillain-BarréSyndrome: A Systematic Review Kate O. Poropatich1, Christa L. Fischer Walker2, and Robert E. Black2 1George Washington School of Medicine and Health Sciences, Washington,
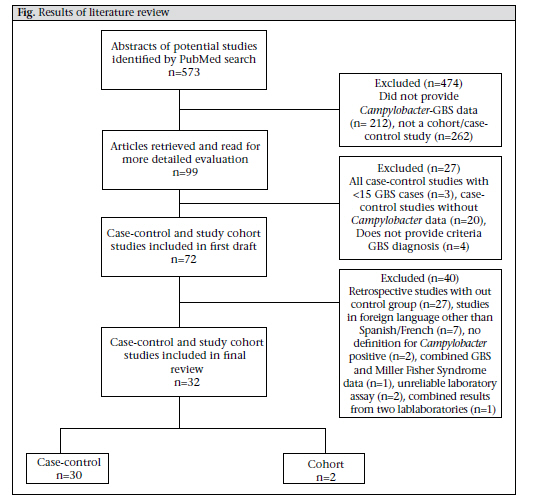
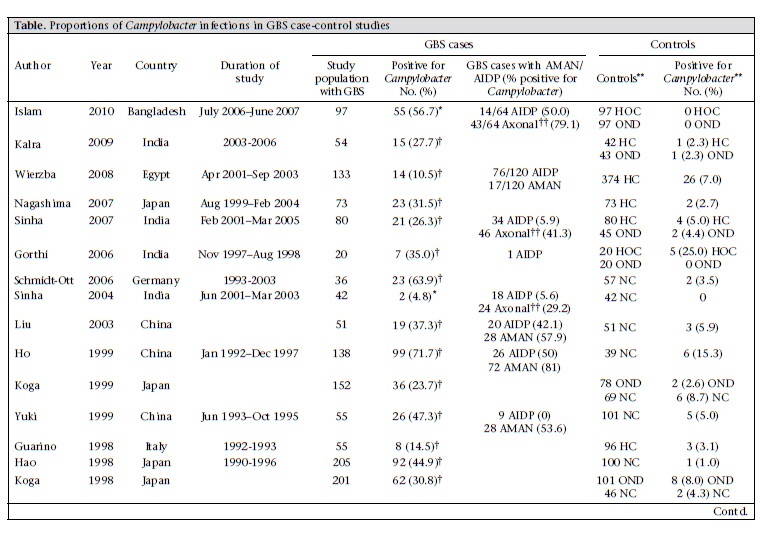
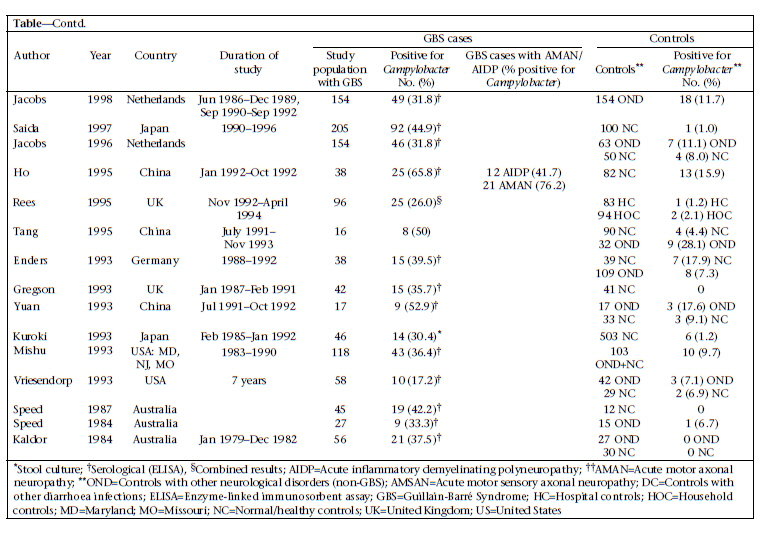
DC, USA and Correspondence and reprint requests should be addressed to: Dr. Christa L. Fischer Walker Department of International Health Johns Hopkins Bloomberg School of Public Health 615 North Wolfe Street, Room E5535 Baltimore, MD 21205 USA Email: cfischer@jhsph.edu Fax: 4109557159 Code Number: hn10071 ABSTRACT Guillain-Barré Syndrome (GBS) is a neurologic disease that causes ascending paralysis and is triggered by a preceding bacterial or viral infection. Several studies have shown that patients with GBS have a recent history of infection due to Campylobacter jejuni. A literature review of published studies that reported rates of Campylobacter infection before or in conjunction with GBS was done. These reported data were used for calculating the proportion of GBS cases who tested positive for Campylobacter compared to the control population and the incidence of GBS among patients infected with Campylobacter. Results of the analysis suggest that 31% of 2,502 GBS cases included in these papers are attributable to Campylobacter infection. Key words: Campylobacter; Campylobacter infections; Guillain-Barré Syndrome; Review literature INTRODUCTION Since the near global eradication of poliomyelitis, Guillain-Barré Syndrome (GBS) has become the most common cause of acute neuromuscular paralysis (1). GBS is an acute neurologic disease driven by autoimmunity and molecular mimicry in which the body stages a cell-mediated and humoral immunological response against peripheral nerve myelin (2). Asbury and Cornblath clinically defined GBS as a progressive motor weakness of more than one limb with low or absent reflexes and no other identifiable cause (3). The global incidence of GBS ranges from 0.4 to 4.0 (median 1.3) cases per 100,000 people annually, occurring slightly more often in adolescents and young adults than in children (2,3). In its acute phase, GBS can cause severe disability and even death (4). A recent systematic review of GBS estimated that 40-70% of all GBS cases are preceded by an acute infectious illness, of which 22-53% are upper respiratory infections and 6-26% are gastrointestinal infections, one of the most common being enteritis due to Campylobacter (5,6). GBS can be classified into demyelinating and axonal subtypes. The demyelinating subtype—acute inflammatory demyelinating polyneuropathy (AIDP)—is characterized by demyelination of neurons whereas demyelination is absent in the axonal subtypes—acute motor axonal neuropathy (AMAN) and acute motor sensory axonal neuropathy (AMSAN) (7). AIDP is common in North America and Europe while AMAN/AMSAN have been more commonly found in studies in China, Japan, and Mexico (2,8,9). Although both the subtypes have been associated with infection due to Campylobacter, it is has been shown that Campylobacter is more widely associated with AMAN (10-12). Campylobacter jejuni was first associated with GBS in 1982 when Rhodes and Tattersfield reported a case of GBS following enteric infection with C. jejuni (13,14). It is difficult to positively associate C. jejuni with GBS because the bacteria are usually eliminated from the body within 16 days of infection and before the onset of neurological symptoms, which normally begin 10 days to 3 weeks after the onset of diarrhoea (1,2). Although Campylobacter is prevalent in most parts of the world, it is not yet routinely diagnosed in rural health clinics. For this reason, many Campylobacter-associated GBS cases may go unrecognized because by the time the person presents with GBS, Campylobacter is no longer present (15,16). Results of a previous overview of literature suggested that Campylobacter infection is responsible for 13-72% of GBS cases; however, this review was not designed to systematically review the literature (2). Thus, we did a systematic review of published studies to estimate the proportion of GBS cases that may be attributed to Campylobacter among persons of all ages and from all regions of the world. MATERIALS AND METHODS We performed a PubMed search of studies published from July 1982 to 28 June 2010 that investigated the relationship between infection due to Campylobacter and GBS. We searched using combinations of the following Medical Subjects Headings (MeSH): ‘Guillain-Barré Syndrome’ and ‘Campylobacter’ and the key words: ‘guillain barré syndrome’, ‘GBS’, ‘acute autoimmune neuropathy’, ‘acute inflammatory polyneuropathy’, ‘acute inflammatory demyelinating polyneuropathy’, ‘AIDP’, ‘AMAN’, and ‘Campylobacter’. We also searched the reference lists of retrieved manuscripts to identify additional studies. We included cohort studies of persons with laboratory-confirmed infection due to Campylobacter who were followed prospectively to assess subsequent GBS cases and retrospective case-control studies that tested for Campylobacter infections among GBS-confirmed cases and non-GBS controls. We excluded case-control studies with fewer than 15 GBS cases and cohort studies, including cases of GBS that developed more than six months after a confirmed infection due to Campylobacter. Studies were included if serum and stool samples were collected during the acute phase of GBS—within 24-48 hours of patient’s admission to hospital and no longer than four weeks after admission. Studies were excluded if they relied on a complement fixation assay (CFA) for the diagnosis of Campylobacter. For case-control studies, the primary outcome considered was laboratory-confirmed presence of infection due to C. jejuni in GBS cases and controls. For cohort studies, the primary outcome considered was the development of GBS in persons with laboratory-confirmed infection due to Campylobacter. Clinical features of GBS were analyzed, and antecedent infections were investigated. Definitions of GBS in the studies were based on currently-accepted criteria for diagnosing GBS (i.e. a progressive, symmetric ascending paralysis with a relative sensory sparing in more than one extremity with hypo- or areflexia) (3,17). Studies were excluded from review if these did not explicitly state or cite their criteria for diagnosis of GBS. Studies were included if these used appropriate microbiological methods (serological assays and stool cultures) for detecting Campylobacter species (13). We reviewed all titles and abstracts to identify eligible studies. Full manuscripts were obtained for potentially eligible studies. Statistical methods For case-control studies, we calculated the median and interquartile range (IQR) for cases and controls and Campylobacter-positive cases and controls. The Microsoft Excel software was used for calculating medians and IQR (18). RESULTS We screened 573 potential studies (Fig.) for inclusion in the review. After applying the inclusion and exclusion criteria, we included two prospective cohort studies (16,19) and 30 case-control studies (Table a & b). In total, case-control studies yielded 2,502 GBS subjects and 3,419 controls. One study took place in the UN-classified (20) least-developed countries (21)—11 in developing countries (10,11,22-30) and 20 in developed countries (12,16,19,31-47). For case-control studies, rates of Campylobacter infection varied among the GBS patients and controls from 4.8% to 71.7% and 0% to 28.1% respectively. The median positivity for Campylobacter among the GBS cases was 35.4% (IQR 28.3-44.9), and among the controls, it was 4.4% (IQR 1.2-8.8), suggesting that 31.0% of the GBS cases may be attributable to a previous infection due to Campylobacter. We identified two cohort studies meeting our inclusion criteria (16,19). In the study by McCarthy et al., 0.03% of Campylobacter cases (n=29,563) developed GBS, which can be expressed as 30.4 cases of GBS per 100,000 cases of C. jejuni-associated infection [95% confidence interval (CI) 13.9-57.8] (16). In a population-based cohort study, Tam et al. found that, of 2,560 persons infected with Campylobacter who received subsequent medical follow-up, three cases of GBS were found, yielding an incidence of 117 per 100,000 cases of C. jejuni (95% CI 0.38-3.63) (19). DISCUSSION In this review, we found 32 studies that met our inclusion/exclusion criteria and measured the association of infection due to Campylobacter with GBS. One of the challenges in determining the incidence of Campylobacter-associated GBS is that many cases of Campylobacter go unreported. For this reason, most studies focus on a sample of persons already diagnosed with GBS and perform retrospective analyses to determine if they were infected with C. jejuni (2). Large cohort studies are ideal for determining the actual incidence of Campylobacter-associated GBS because they identify Campylobacter cases at the time of active infection and are, thus, less likely to miss a Campylobacter infection because of poortiming.Unfortunately,thesestudiesarerare. We used a standard definition for GBS to minimize differences in diagnostic criteria among studies (3). These are the internationally-accepted criteria currently used for diagnosing GBS and helped control for study heterogeneity. One limitation of this analysis is controlling for heterogeneity among studies that met our inclusion criteria. Depending on whether studies used serologic assays or stool samples for the diagnosis of Campylobacter, different positive values could be shown. Serology is the preferred mechanism of detection because Campylobacter-specific antibodies can be detected in serum of the patient for an indefinite length of time compared to Campylobacter antigens in stool samples, which are cleared, on average, 16 days after infection. Thus, there could be many false-negative stool cultures in GBS patients in studies that rely solely on stool culture for the diagnosis of Campylobacter (48). Additionally, there is no serological test that is specific for Campylobacter infection as far back as in the two months before the onset of GBS; so, there is potential misclassification of exposure in retrospective serological studies, which comprise the bulk of the case-control studies considered in this analysis. Variations in serological assays could also affect results of study; antigens used and endpoints for positivity often vary with different assays (48). It is preferable that studies using serology to adhere to strict criteria for recent diagnosis of Campylobacter (i.e. positive ELISA for at least 2 classes of antibodies) to avoid false-positive results. However, we did not specifically exclude studies on the basis of the number of Campylobacter-specific antibodies they used, only if they did not explicitly state what their standard was for seropositivity of Campylobacter. Finally, we did not control for improving technologies for the detection of Campylobacter over the years of the studies, which could also confound the comparison of results from older studies with more recent studies. Another limitation to take into consideration is the overestimation of Campylobacter infectivity in control groups. While serological evidence of Campylobacter infection is likely indicative of recent infection in GBS cases due to the strong temporal link between Campylobacter infection and GBS, this is not the case for controls. If controls test positive, there is no way of knowing how long ago their infection occurred because antibody titres remain elevated for an extended period. The median value of 4.4% for Campylobacter positivity in the control group in our study may represent individuals with infection due to Campylobacter beyond two months and, thus, could be an overestimate of active Campylobacter infections in the general population. If this were the case, the prevalence of Campylobacter-associated infections would actually be greater between cases and controls. The instance of heterogeneous control populations in the studies is an additional limitation of this analysis. Some studies included controls with other neurological disorders with preceding C. jejuni-associated infection as high as 28.1%, which is considerably higher than that has been observed in other control groups. Selection of controls in the study could alter the difference in Campylobacter positivity between GBS cases and controls, depending on whether the control groups’ risk of exposure was the same as the GBS group or different. For instance, household controls could have a risk comparable to GBS cases, and high rates of seropositivity among these controls could reflect transmission of Campylobacter from cases and controls, along with unhygienic living conditions in developing countries. We were also unable to search the Chinese medical literature databases or review Chinese language papers identified in PubMed. We recognize that these data may exist and would better help us understand the associations of GBS with Campylobacter in China. While infection due to Campylobacter is not normally associated with high rates of mortality in developed countries, 4-15% of patients with GBS may die within the first year after onset (49). C. jejuni has been identified as a potential predictor of poor outcome in persons suffering from GBS for inducing a more severe autoimmune response and greater axonal damage (2). Some studies report that preceding Campylobacter infection can increase the severity of GBS in patients, i.e. death, mechanicalventilation,etc.(2).Thisisproblematic in poorer countries that usually have increased frequencies of infection due to Campylobacter because persons who develop GBS may have limited access to healthcare and treatment required for GBS. For these reasons, the appropriate measures must be taken to reduce the incidence of Campylobacterassociated GBS. This can be achieved through reducing the frequency of Campylobacter cases by improving sanitation, preventing the faecal-oral routes of transmission of Campylobacter. REFERENCES
Copyright 2010 - Journal of Health Population and Nutrition The following images related to this document are available:Photo images[hn10071t1b.jpg] [hn10071t1a.jpg] [hn10071f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}