 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
African Health Sciences, Vol. 1, No. 1, December, 2001, pp. 83-89 Rapid detection of Mycobacterium avium subsp. paratuberculosis from cattle and zoo animals by Nested PCR 1Joseph Erume, 2Joachim Spergser and 2Renate Rosengarten 1Faculty of Veterinary Medicine, Makerere University, P.O.Box 7062, Kampala, Uganda. Correspondence: Dr. Joseph Erume, Faculty of Veterinary Medicine, Makerere University, P. O. Box 7062 Kampala, Uganda. Tel: 256(41)533002; Fax: 256(41)534336. Email: erujoseph@yahoo.com Code Number: hs01023 ABSTRACT Paratuberculosis, caused by Mycobacterium avium subsp. paratuberculosis, a suspect causative agent of Crohn's disease in man, is an emerging disease of international proportions affecting all ruminants. Early stage detection of Mycobacterium avium subsp. paratuberculosis infection would accelerate progress in control programmes. Despite new molecular approaches the standard diagnostic test for this disease is at present still the time consuming classic isolation procedure. Therefore, alternative diagnostic tests such as PCR, are needed for quick detection of infected animals. In this study, the conventional enrichment and isolation procedure and two IS900-based PCR methods for detection of Mycobactrium avium subsp. paratuberculosis in clinical samples from zoo animals and cattle were compared. A total number of 48 different clinical specimens obtained from animals suspected of having paratuberculosis were examined. The samples included faeces (n = 15) and organ tissues (n = 33). Of the faecal specimens two were identified as positive by nested PCR, whereas none was positive by single PCR or by culture. 28 organ specimens were found positive by culture. Mycobactrium avium subsp. paratuberculosis DNA was detected by nested PCR in 82% of the organ specimens identified positive by culture (23 samples) as opposed to 57% by single PCR (16 samples). Nested PCR also identified two positive samples that were not detected by either culture or single PCR. These findings show the great potential of nested PCR as a useful tool for the rapid diagnosis of paratuberculosis in animals. INTRODUCTION Paratuberculosis or Johne’s disease is a chronic progressive infectious disease that affects all categories of domestic and wild ruminants including cattle, goats, camels, buffaloes and farmed dear 1,2. It is caused by Mycobacterium avium subsp. paratuberculosis, a small, fastidious acid-fast bacterium. This organism is also suspected to be a potential causative agent of Crohn’s disease in humans 3, 4. The disease occurs throughout the world and is responsible for considerable economic losses. There is no therapy and it invariably leads to the death of the affected animal5. Control of the disease in ruminants is dependent on the early detection and culling of infected animals. Attempts to the control are, however, severely hampered by inadequate diagnostic procedures. At present, the standard diagnostic procedure involves culture of the organisms from faeces 6,7. The problem with the culture procedure is that it is time consuming and requires 8-16 weeks of incubation and use of specialised culture medium 8. Despite decontamination steps, cultures are often lost because of contamination. In addition, clinically infected animals may shed organisms sporadically during disease and therefore false negative results may occur 9. Furthermore, though the slow growth rate and the dependence on exogenous mycobactin for in vitro growth are characteristic for Mycobacterium avium subsp. paratuberculosis, these are not enough to differentiate it from other mycobacteria in particular Mycobacterium avium subsp. avium and Mycobacterium avium subsp. silvaticum10. Due to the limitations associated with the culture method, a number of other tests have been developed to aid in the diagnosis of paratuberculosis 6,8,11. The technique right now that is the most focus of attention is the polymerase chain reaction (PCR). The potential value of the PCR in diagnosing Mycobacterium avium subsp. paratuberculosis infections has been recognised for some time and this technique has been applied in a variety of clinical samples 12. A break through in the use of the PCR for diagnosis of paratuberculosis was the detection of IS900, a repetitive DNA insertion element unique to Mycobacterium avium subsp. paratuberculosis. IS900 can be specifically detected by PCR 9,13,14. The PCR technique offers specific and rapid detection of Mycobacterium avium subsp. paratuberculosis, however, reduced levels of sensitivity have been encountered when it has been applied on clinical samples, and these reductions have been attributed to the presence of inhibitors 9,12. Although inhibitors undoubtedly are involved, the poor sensitivity is also likely a result of inefficient amplification of DNA. This follows a result of findings that sensitivity of amplification may be enhanced by nested polymerase chain reaction, a two-step amplification procedure 15. The purpose of this study was to assess the potential of nested PCR for the rapid detection of Mycobacterium avium subsp. paratuberculosis in clinical samples from animals and compare its performance with single PCR and bacteriological culture. METHODS Samples
Ziehl-Neelsen Staining
Culturing of bacteria from clinical samples
Culturing from tissues was carried out on HEYM after decontamination with HPC as for faecal samples. First 1 g of tissue was weighed and homogenised in 0.5 ml sterile distilled water using a mortar and pestle. The homogenate was then transfered to a 50 ml centrifuge tube. 25 ml of 0.9% HPC was added to the homogenised material, shaken, and the material allowed to stand for 30 minutes. After settling, the cellular fraction (minus tissue fragments) was transfered to a second 50 ml centrifuge tube. Decontamination of the sample was carried out overnight at laboratory temperature. This was followed by centrifugation at 1700 x g for 20 minutes. The supernatant was discarded and 0.1 ml of sediment per tube was inoculated on HEYM after resuspending the pellet in 0.5 ml sterile water. Inoculation and incubation of samples was done as for faecal samples. Extraction of DNA from faecal samples
Isolation of DNA from tissue samples
DNA extraction and subculture of colonies Analysis of clinical samples with Single PCR
Analysis of clinical samples with Nested PCR
To reamplify the DNA, 1 µl of each of the first amplification products was transfered to tubes containing the same reaction mixture described above except that oligonucleotide primers para 1 and para 4 were replaced by oligonucleotides para 2 ( 5' GCC GCG CTG CTG GAG TTG A 3') and para 3 (5' AGC GTC TTT GGC GTC GGT CTT G 3'), which recognise sequences contained within the sequence amplified by para 1 and para 4 and an amplicon target of 210 base pairs 15. The samples were subsequently amplified for an additional 35 cycles as described above. Agarose gel electrophoresis
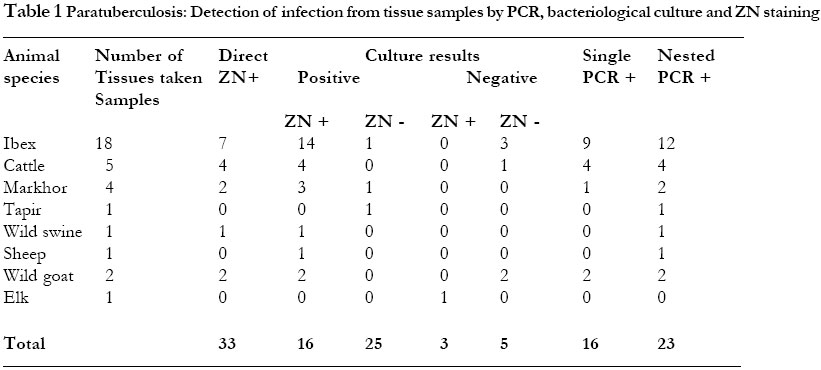
RESULTS We have tested 48 clinical specimens from animals suspected of having paratuberulosis. Of these samples 33 were tissues (ileum, mesentric lymph nodes and liver) and 15 were faeces. These specimens were obtained from zoo animals including ibex, markhor, elk, tapir, waterbuck, wild goeat, wild swine, bison and from sheep and cattle. Detection of infection from tissue samples28 of the 33 tissue samples were found to be culture positive. Colonies compartible with M. avium subsp. paratuberculosis were first noted at 8 weeks of incubation. But the majority of growths were seen from 10-16 weeks of incubation. 11 samples grew colonies other than M. avium subsp. paratuberculosis. Of these samples, one showed colonies within two weeks while in the remaining 10 samples contaminant colonies were observed at 4 weeks of incubation. In all these samples, contaminants were seen in all the 4 tubes inoculated per sample. Later colonies compartible with M. avium subsp. paratuberculosis were also seen in the 11 samples. Suspect colonies were characterised by ZN staining and confirmed by PCR as of M. avium subsp. paratuberculosis . Furthermore, isolates were found to be mycobactin dependent when subcultured. The identity of contaminants was not determined. The 33 different clinical tissue samples were tested by single PCR and 16 of the 33 tissue specimens were found positive. When nested PCR was applied on the DNA from the clinical samples, 23 were found positive. None of the 5 tissue samples that were found negative by culture were positive by nested PCR. The details of results of culture, ZN staining and PCR on tissue samples are presented in Table 1.
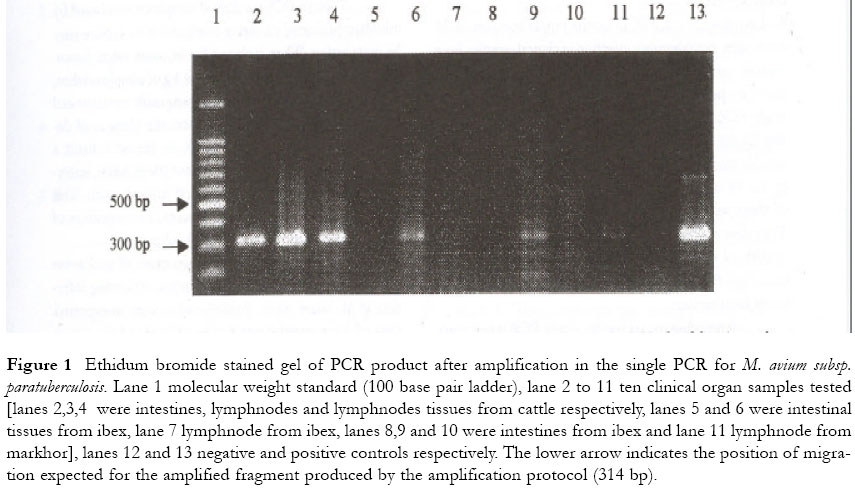
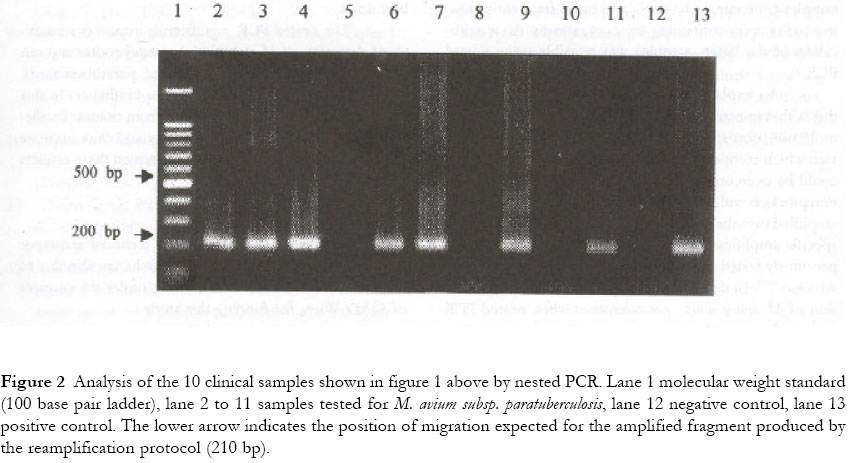
One of these samples was found to be negative by culture and by PCR and was therefore classified as false positive. In this study, direct microscopy showed a detection rate of 57% (16/28) similar to that of single PCR. 10 clinical organ samples tested by both methods of PCR are shown in figure 1 (single PCR) and figure 2 (nested PCR). Figure 1 Ethidum bromide stained gel of PCR product after amplification in the single PCR for M. avium subsp. paratuberculosis. Lane 1 molecular weight standard (100 base pair ladder), lane 2 to 11 ten clinical organ samples tested [lanes 2,3,4 were intestines, lymphnodes and lymphnodes tissues from cattle respectively, lanes 5 and 6 were intestinal tissues from ibex, lane 7 lymphnode from ibex, lanes 8,9 and 10 were intestines from ibex and lane 11 lymphnode from markhor], lanes 12 and 13 negative and positive controls respectively. The lower arrow indicates the position of migration expected for the amplified fragment produced by the amplification protocol (314 bp). Figure 2 Analysis of the 10 clinical samples shown in figure 1 above by nested PCR. Lane 1 molecular weight standard (100 base pair ladder), lane 2 to 11 samples tested for M. avium subsp. paratuberculosis, lane 12 negative control, lane 13 positive control. The lower arrow indicates the position of migration expected for the amplified fragment produced by the reamplification protocol (210 bp). Overall, 30 clinical samples were identified as positive, with culture showing a detection rate of 93% (28 samples), nested
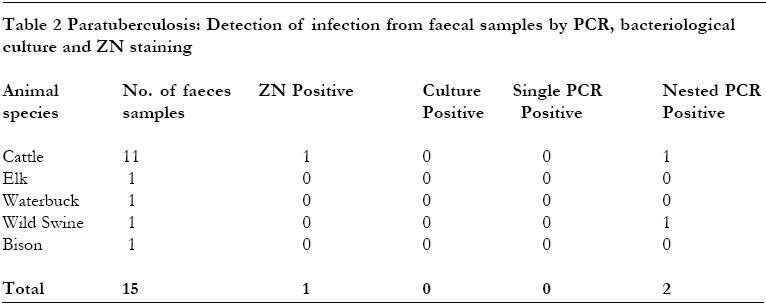
PCR 83% (25 samples) and both single PCR and ZN a rate of 53% (16 samples). We have tested nested PCR for the rapid detection of M. avium subsp. paratuberculosis infection in clinical samples from animals suspected of having paratuberculosis and compared its performance with bacteriological culture and single PCR. Bacteriological culture is currently the main stay for the diagnosis of this disease but its time consuming. M. avium subsp. paratuberculosis was isolated from 28 of the 48 clinical samples tested (Table 1 and 2). Sixteen of these were identified by single PCR test as infected. The infected samples detected by single PCR test showed growth of colonies at 8 weeks of incubation implying that single PCR is able to detect clinical samples with high levels of infection. According to our results, single PCR is not sensitive enough as a diagnostic test for clinical samples. This low sensitivity of single PCR has been reported previously and was attributed to the presence of inhibitors of the amplification 9,12. Studies on Mycobacterium tuberculosis have, however, found that this lower sensitivity of the standard amplification protocol when applied on clinical samples is neither solely due to presence of inhibitors nor to extraction of mycobacterial DNA-. It was found that when a standard amplification protocol is used, the technique can detect DNA from Myobacterium tuberculosis in samples containing >100 CFU/ml but is frequently negative for samples containing fewer organisms. An amplification of the latter samples was possible using nested PCR 21. An explanation that has been put forward for this is that in reactions containing low numbers of target molecules, non-specific amplification products are generated which compete with the mycobacterial DNA 19. This could be overcome by reamplifying the initial amplification products with primers which recognise the inefficiently amplified mycobacterial DNA but not recognise the nonspecific amplification products. This approach has been previously tested for detection of M. avium subsp. paratuberculosis 15,18. In this present study, the sensitivity of detection of M. avium subsp. paratuberculosis when nested PCR was used was evidently superior to that obtained when single PCR was applied. Interestingly, Nested PCR detected 2 positive faeces samples which even culture had not identified. The nested PCR detection rate of 82% from the clinical organ samples is somewhat disappointing because it showed a lower sensitivity compared to bacteriological culture. The main reason for this lower sensitivity of nested PCR in this present study was its failure to amplify 5 tissue samples that were found positive by culture. The failure to amplify these samples could be attributed to the presence of inhibitors of Tag polymerase in the original samples. Though PCR on clinical samples is cumbered by inhibitor problem, its direct comparison to culture may be quite unfair. When isolating for M. avium subsp. paratuberculosis from clinical samples about 1 g of sample is taken, decontaminated and organisms subsequently concentrated before culture. This greatly enhances the chances of detecting the positive cases. For PCR on the other hand, a comparatively smaller amount of sample is taken, implying that few organisms are targeted to begin with. This may be another reason for the apparent low sensitivity of PCR as compared to conventional culture. On the other hand, the two cases of wild swine that responded to IS900 PCR primers, indicating infection of M. avium subsp. paratuberculosis were unexpected. One of these samples was a piece of liver and the other a fecal sample. On culture, the tissue sample showed organisms other than M. avium subsp. paratuberculosis within 4 weeks of incubation. Colonies compatible with Johne's disease bacilli were observed later after 8 weeks of incubation and on tubes with mycobactin J only. This was probably a case of a mixed infection. Previously M. avium subsp. paratuberculosis was thought to infect only ruminants, however, increasingly infection is being detected in a variety of non-ruminants as well 22. This finding has important epidemiological implications in the control of paratuberculosis. The nested PCR significantly improves sensitivity of detection of M. avium subsp. paratuberculosis and can be useful for the quick diagnosis of paratuberculosis. However, PCR inhibitors are a major hindrance. In this study we used crude DNA extracts from tissues. To alleviate the problem of PCR inhibitors and thus improve the sensitivity, a phenol chloroform step on these extracts could be of help. ACKNOWLEDGEMENTWe thank Martina Wukovits for the technical assistance she rendered towards this work. Thanks are also due to the Federal Government of Austria, under the auspices of ÖAD, Wien, for funding this study. REFERENCES
Copyright 2001 - Makerere Medical School, Uganda The following images related to this document are available:Photo images[hs01023t1.jpg] [hs01023f1.jpg] [hs01023f2.jpg] [hs01023t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}