 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
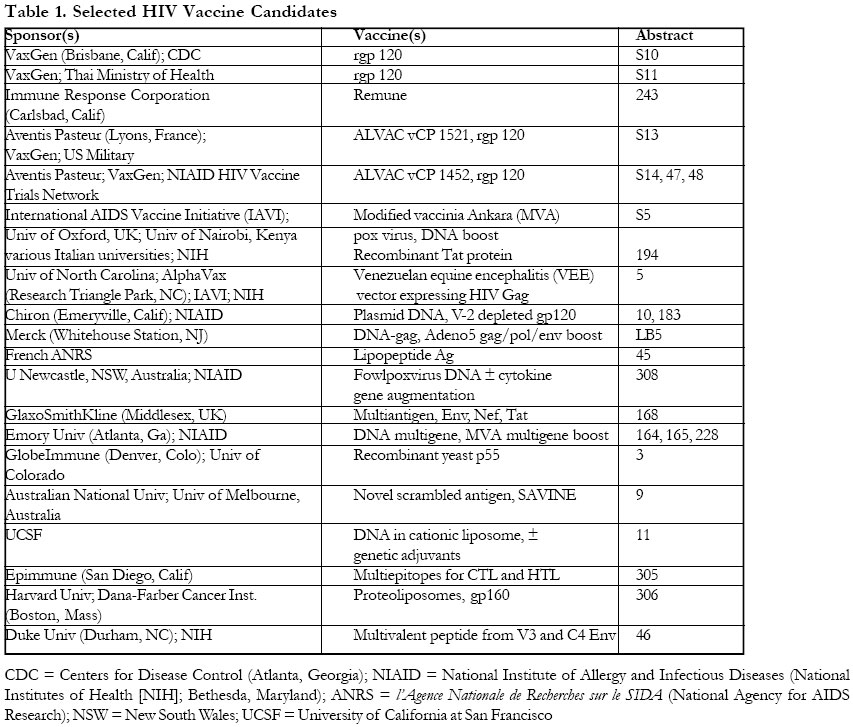
African Health Sciences, Vol. 1, No. 1, December, 2001, pp. 99-105 PRACTICE POINTS AIDS Vaccine 2001: Looking to the Future 1Ronald T. Mitsuyasu 1Associate Professor of Medicine, University of California Los Angeles, and Director of the UCLA CARE Center, Reprinted with kind permission from Medscape HIV AIDS (http://w.w.w.medscape.com/Medscape/HIVjournal/2001/ Correspondence: Ronald T.Mitsuyasu, MD Associate Professor of Medicine University of California Los Angeles and Director of the UCLA CARE Center, UCLA Medical Center, Los Angeles Code Number: hs01026 NTRODUCTION AIDS Vaccine 2001, a new addition to the international conference calendar that will undoubtedly become a biannual event, was designed to provide a setting for sharing the latest basic, clinical, and public health data relevant to AIDS vaccine development and to facilitate international and interdisciplinary collaboration in the field of AIDS vaccinology. The 3-day meeting in Philadelphia provided considerable information on the preclinical development and early clinical evaluation of several vaccine candidates, and ample opportunity for discussion on AIDS vaccine and immunotherapy study implementation. The overall impression is that a great deal of effort and considerable expertise is now being directed towards dissecting the immunologic and virologic components of protective immunity against HIV and towards the development of novel immunotherapeutic approaches to the prevention of HIV infection. This effort is in no small part due to the worldwide attention being given to the devastating effects of HIV and AIDS in resource-poor, developing countries of the world, and to the realization that treatment of HIV in and of itself is unlikely to contain the spread of this epidemic. In an insightful overview of recent progress in the treatment and prevention of HIV worldwide, Anthony Fauci, MD,[1] Director of the National Institute of Allergy and Infectious Diseases (NIAID), reminded the audience that more than 58 million people worldwide have been infected with HIV since the beginning of the pandemic and that an estimated 5.3 million people, most of them living in developing countries, were infected with HIV in the year 2000 alone. Over 3 million deaths due to AIDS occurred in the year 2000, and cumulatively over 21.8 mil lion people have died of AIDS-related complications since the initial recognition of this disease. “Historically, vaccines have provided safe, cost-effective and efficient means of preventing the illness, disability, and death from infectious diseases. The development of a safe and effective vaccine for HIV infection is an essential goal of AIDS research and a necessary tool to bring the HIV epidemic under control,” said Dr. Fauci. With funding for HIV vaccine research increasing more than 6-fold since 1990 to an estimated $356 million for fiscal year 2002, work on developing new HIV vaccine strategies and on developing infrastructure for the conduct of necessary clinical trials has rapidly expanded in the last few years. In concert with the increase in scientific and clinical efforts in this area have come several key scientific advances in the understanding of HIV-specific immunity, including recognition of the importance of generating broad-based and long-lasting HIV-directed cytotoxic T lymphocyte (CTL) responses as well as broad neutralizing antibodies against free virus, especially in the early phases of infection. In addition, a better understanding of the importance of HIV clade and strain diversity and of the mechanisms of escape of virus replication from immune control is helping to define some of the potential limitations for developing effective protective immunity against HIV. Several recent successful animal challenge experiments after SIV- and SHIV-specific vaccinations have generated much enthusiasm and have led to great hopes that a protective vaccine for HIV may soon be on the horizon. Nevertheless, given the differences between humans and other primates and between SIV and HIV, extrapolation from these early animal studies must not be overblown. I believe the mood of many of the participants at the end of this conference might best be summed up as “cautious optimism”: cautious because of the formidable challenges that remain in better understanding the underpinnings of HIV's interaction with the immune system and escape from immune control, and how best to exploit these findings to treat and control this disease; optimism because of the increasing scientific, social, and political efforts now being directed at these problems and especially the development of new vaccines and immunotherapies. As one involved in the care of patients with HIV infection since almost the beginning of the epidemic, it is gratifying to see the renewed interest in host defenses against HIV as both a meansof treating and preventing this infection. While it is impossible to provide a comprehensive review of all significant reports from this conference, I will aim to focus on afew of the most important, clinically relevant topics and presentations. Principles of HIV-Specific ImmunityProtective immunity against HIV involves both humoral and cellular immunity. Specifically, protection requires neutralizing antibodies directed to various epitopes expressed by HIV itself as well as cellular immune responses, particularly CTLs targeted to various epitopes expressed on the surface of HIV-infected cells. The CTL response is triggered by HIV-specific T-helper lymphocytes (THLs) and by the generation of cytokines, both of which are produced from activated CD4+ cells in response to the presentation of HIV antigens by antigen-presenting cells (APCs) such as dendritic cells and macrophages. However, in most cases of HIV infection the rapid loss of HIV-specific THLs and functional abnormalities in a variety of other immune cells ultimately lead to the establishment of chronic infection and a level of ongoing viral replication (the set point) which, if untreated over time, results in further progressive loss of immune function. The goal for a preventive HIV vaccine is to generate both humoral and cellular immunity against HIV in the host before exposure to the virus. Following initial exposure to HIV, the generation of cellular immune responses against HIV may take a while to develop, and therefore neutralizing antibodies against free virus are important to dampen initial viral spread. Subsequently, generation of HIV-specific THL and CTL responses becomes important in removing HIV-infected cells from the host and in controlling further activation and spread of the virus once established in the host. Thus, both arms of the immune system are important in the immunologic control of HIV infection.[2] Identifying which epitopes of HIV are most critical in establishing infection or, conversely, which epitopes should be targeted for the development of cell-mediated and humoral immune responses to control HIV, is a major concern in vaccine development. Due to the considerable genetic diversity among HIV clades and strains and the rapidity of viral mutation, most efforts to date have been targeted at conserved epitopes in the gag or pol gene for CTLs and in the V3 loop area of the HIV Env for neutralizing antibodies. This approach was taken because of early findings that abrogation of CD8+ CTLs directed against these major conserved epitopes resulted in loss of protective immunity and rapid progression of SHIV disease in monkeys. Likewise, most protective neutralizing antibodies found in patients with long-term nonprogressive HIV infection were directed against conserved regions of Env and some of the regulatory proteins. However, studies of immunogens that generate solely humoral immune responses to conserved Env epitopes have failed to show protection in animal challenge studies and have been generally ineffective in providing sufficient immune enhancement to control infection in chronically infected individuals. CTL-directed vaccines have been much more difficult to develop, as they depend on effective presentation of antigens in a biologically appropriate format, such as in association with appropriate major histocompatibility (MHC) antigens that generally require processing within cells such as can be achieved with live viral vectors. These vaccines are also dependent on the appropriate functioning of the APCs, THLs, and necessary cytokines to help generate the response. Attempts to accomplish this have included (1) the incorporation of the genes for the important epitopes in live viral vectors that could infect T cells and thereby present the important epitopes on the cell surface in association with MHC antigens in a natural way, and (2) using immune adjuvants such as cytokines or chemicals that can potentiate the cellular immune response. A third approach would be to associate the immunogens with APCs such as dendritic cells, which could then present the important epitopes to helper and cytotoxic T cells. Whole killed or inactivated, replication-incompetent HIV vaccines are yet another approach that would present a broad array of HIV antigens to THLs and/or CTLs and thereby may obviate concerns about whether the appropriate genes and epitopes have been selected. Mutations in the viral genes for these antigens might result in immunologic escape, especially if only a few antigens are targeted in the vaccine. In addition, mutations in viral genes coding for the binding region of MHC class I proteins, which may also result in viral escape from immunologic control, have been described.[3,4] A final concern is whether differences between HIV clades may be sufficiently important to require the development of clade-specific and perhaps subtype-specific vaccines for use in different regions of the world, in case crossclade immunity against HIV should prove not to be sufficiently potent to prevent viral infection or to suppress viral replication. Rationale for Therapeutic VaccinesThe concept of therapeutic HIV vaccination is based on the premise that generation of HIV-specific immune responses in individuals who are already infected may help to suppress viral replication, and may thus allow reduction in the intensity of antiretroviral therapy or even its discontinuation for some period of time. Bruce Walker, MD,[3] from Massachusetts General Hospital discussed some of the reasons for optimism for the development of therapeutic vaccines, as well as some of the obstacles to their implementation. Studies in acute HIV infection have demonstrated that treatment with antiretroviral therapy soon after infection may preserve HIV-specific host immunity and that transient control of viral replication may be achieved in some of these individuals after cessation of therapy.[5,6] Long-term follow-up of 14 such subjects with acute infection treated with highly active antiretroviral therapy (HAART) who then underwent treatment interruption demonstrated that 6 maintained persistent control of HIV viremia out to day 600. Other individuals, however, experienced recrudescence of viral replication, in many cases as late as 500 days or longer after stopping therapy. The considerable heterogeneity in the time course of these responses suggests that in some cases virologic escape - perhaps due to expansion of viral diversity and escape from immunologic control - or the gradual loss of protective immunity may have occurred. Dr. Walker reported that the most immunodominant of the CTL epitopes in these patients was directed to Vpr and to p17.[7] It is possible that in such individuals, treatment with CTL-inducing therapeutic vaccines may allow control of viral replication for prolonged periods, while avoiding the development of the viral mutations that may be expected if endogenous HIV replication is permitted, eg, during structured treatment interruptions (STIs). Potential obstacles to the use of therapeutic vaccines in HIV include:
Nevertheless, Dr. Walker emphasized the importance of continuing to study therapeutic vaccination strategies, both because of the importance of evaluating the concept of possibly enhancing the host's ability to control viral infection endogenously, and because of the many difficulties inherent with current long-term use of antiretroviral therapy. Studies of Therapeutic vaccinationSeveral studies of therapeutic vaccination approaches were presented at a poster session on this topic. In a study by Lindenburg and colleagues[8] from Amsterdam, a vaccine comprising HIV-1 p17-p24:Ty virus-like particles (p24-VLP, British Biotech) was administered to 74 asymptomatic HIV-infected individuals in a phase 2 trial conducted in 1993-1994. No differences were seen in changes in CD4+ cell count, use of antiretroviral therapy, or AIDS progression rates between vaccinated and unvaccinated individuals. However, this study was conducted in the pre-HAART era and many of the patients received less than optimally immunogenic doses of vaccine. In an open-label pilot study in chronically infected individuals on HAART with undetectable plasma HIV-1 RNA levels and CD4+ cell counts > 350 cells/mm3, patients received 6 HIV lipopeptides (3 Nef, 2 Gag and 1 Env) in mix micelles.[9] Patients received 3 injections performed 3 weeks apart, and HAART was then interrupted at week 24. Viral rebound was observed in all patients after a median delay of 2 weeks, with a peak viral load at week 3 followed by a lower plateau period. It was noted that drug-resistant strains of virus were detected at the time of viral rebound in several of these patients after treatment interruption. In a small nested study of a larger controlled trial of Remune + Incomplete Freunds Adjuvant (IFA) vs IFA alone control in chronically infected individuals, it was noted that the slope of the initial rise in plasma HIV1 RNA after treatment interruption was somewhat slower in the Remune recipients compared with the control group (0.16 vs 0.21 log10 copies/mL per day).[10] Although the lymphoproliferative (LPA) response to p24 antigen did not appear to correlate with either of the peak or postpeak viral load changes after treatment interruption, it appeared that the frequency of cells producing interferon-gamma in response to several HIV proteins was significantly increased in the Remune-treated group. In a study by Jin and colleagues[11] from the Aaron Diamond AIDS Research Center in New York, 14 individuals were treated with HAART within 120 days of acquiring HIV infection. These patients also received canary pox vCP1452 and recombinant gp160 vaccination on days 0, 30, 90, and 180 of the study. A total of 13 of the 14 patients completed their vaccination, and all 13 generated antibodies to gp160. Eight of the 14 had LPA responses to gp160, and 8 of 14 had LPA responses to p24. CTL responses as measured by interferon-gamma expression to 1 or more of the env, gag, pol, or nef gene products were observed in 7 of the 14. Overall, 70% of these individuals had some degree of cellular immune response to HIV. Vaccinated patients who underwent a treatment interruption 2 weeks after the last dose of vaccine were compared with a historical control group of unvaccinated patients undergoing treatment interruption following HAART therapy during acute HIV infection. Both groups had decreases in their CD4+ cell counts, and all had viral rebound relatively rapidly within the first 22-27 days. While the numbers are small, there did appear to be some correlation between the proportion of interferongamma producing CD8+ cells and the level of viral rebound. Moreover, 6 of the 11 vaccinated patients who interrupted treatment subsequently achieved and maintained a > 1 log10 copies/mL reduction in plasma viremia from their post-discontinuation peak levels, compared with 1 of 5 unvaccinated historical control patients. This study lacked concurrent controls and involved relatively small numbers of patients, but it does suggest that in those patients with acute HIV infection who generate a good cellmediated response to therapeutic vaccines, some degree of virologic suppression may occur upon stopping therapy. Another study using the same ALVAC vCP 1452 vaccine with or without 3 STIs followed by an analytic treatment interruption (ATI) compared with a control group who receive treatment with HAART alone followed by ATI, is currently in progress in the AIDS Clinical Trials Group (ACTG; study A5068). A similar randomized controlled study of ALVAC vCP 1452 with or without IL-2 is also being performed in chronically infected individuals with fully suppressed viremia and CD4+ cell counts > 350 cells/ mm3 on their first HAART regimen within the ACTG (study A5024). HIV Vaccine CandidatesWithin the last 2 years, many potential vaccine candidates have been developed and are in various stages of preclinical and early clinical evaluation. About 25 of these vaccines were discussed to some degree at this meeting. To date, however, only 1 preventive vaccine - the VaxGen rgp120 vaccine - has entered phase 3 clinical trials. These studies[12,13] now under way in the United States, Canada, The Netherlands, and Thailand follow earlier studies that demonstrated production of neutralizing antibody responses to HIV gp120. A large US Army/Royal Thai Army collaborative study of a prime/boost vaccine strategy, using ALVAC vCP 1452 followed by VaxGen rgp120, will begin soon in Thailand.[14] Table 1 lists some of the vaccine candidates discussed at this meeting, with the program's abstract numbers noted for reference. Most of these vaccine candidates have been shown to generate cell-mediated immunity responses and/ or antibody responses to varying degrees in various animal models. While it is difficult to assess the relative merits of these various vaccine candidates, the large number of vaccines under evaluation suggests that some of these candidates will likely advance to early clinical testing in the not-too-distant future. The vaccines that appear to be furthest along in their clinical evaluation include the canary pox ALVAC vCP vaccines (vCP 205, vCP 1433, vCP 1521, and vCP1452); the modified vaccinia Ankara (MVA) gag-pol or MVA gagpol-env vaccines; the Merck plasmid DNA gag and the Merck adenovirus 5 vector consensus gag-pol-nef vaccine; and the French ANRS lipopeptide antigen vaccines. Due to the preliminary nature of much of the data presented at this meeting and the lack of human clinical data for most of these vaccines, the reader is referred to the abstracts and to the presenters for more information regarding details of the individual vaccines. Lessons From Animal StudiesBy far the most encouraging data from this meeting came from recently presented and published results of SHIV challenge studies in vaccinated primates, which were eloquently reviewed by Norman Letvin, MD,[15] from Harvard Medical School and the New England Deaconess Hospital, Boston, Massachusetts. Dr. Letvin reviewed recent studies in macaques immunized with plasmid DNA vaccines or DNA with pox virus vector vaccines, describing the marked decreases in viral set point and apparent immunologic control of virus replication observed after challenge with SHIV. These protective effects appeared to be closely correlated with the generation of CTL and neutralizing antibody responses to the immunogens. These proof-of-concept animal studies were designed to demonstrate the immunogenicity of vaccines and their clinical correlation with viral protection. In these studies, animals were not protected from infection, but active viral replication and mortality were significantly reduced. CD4+ cell counts were maintained along with partial containment of virus replication in all cases, with durability of effect lasting out to 1.5 years.[15,16] Animals receiving plasmid DNA vaccines (gag, pol, env) with cytokine augmentation (IL-2/ Ig protein or plasmid) appeared to have greater viral suppression and longer survival than those given vaccine alone.[17,18] Durability of response and suppression of viremia exceeds 1.5 years suggesting that perhaps both the immune response and the suppression of virus may be long-lasting. These studies suggest that a vaccine that generates HIV-specific CTL responses in humans could similarly protect against viral replication and HIV disease progression, although it may not necessarily prevent infection with HIV. Similar studies using live pox viral vector vaccine (eg, rMVA-gag, pol ± env) likewise showed good CTL responses and protection from SHIV rechallenge.[17,19,20] Here again, the presence of the env gene in the immunogen added to the protection seen from SHIV challenge.[16,19] In addition, studies of the recombinant adenovirus vector expressing only SIV Gag likewise have been shown to generate potent CTL responses and can protect against CD4+ cell count loss and disease progression after SHIV challenge.[21] Nevertheless, some additional questions remain. For example, animals with pre-existing immunity to the viral vector may show less immune effectiveness from the vaccine; is the level of protection then diminished? This concern clearly has implications for humans, since previous infection with the virus from which the viral vector is prepared (eg, previous vaccinia or adenovirus infection) may inhibit the establishment of effective protective immunity. It also remains to be determined whether the lower viral load seen after infection of vaccinated subjects results in a decreased risk of viral transmission. The take-home message from these animal challenge studies is that vaccination with multiple HIV epitopes, especially if introduced using live viral vectors, with or without boosting and with or without cytokine augmentation, can generate long-lasting protective immunity. Even if not completely protective against primary infection, these vaccines may reduce the viral set point, preserve CD4+ cells, and delay or prevent clinical disease progression and mortality. If similar results can be demonstrated in humans, we will be well on our way towards an effective vaccine strategy. While it is clear that humans are different from monkeys, these primates are our closest known nonhuman relatives and therefore similar biologic responses may be anticipated. The reader is again referred to specific abstracts from the meeting or recent publications[16,18] for more details. Challenges of Clinical StudiesThe development of an effective, clinically beneficial, widely accessible preventive vaccine for HIV is clearly more complex than simply designing a vaccine that is safe and effective in generating an immune response. These issues were addressed in several symposia at this meeting, and discussed on many levels. From a clinical trials standpoint, demonstration of safety is paramount as healthy individuals are involved, and therefore the risk of acquiring HIV infection must be weighed against any potential toxicities from the drug. Establishing a clinical trials infrastructure and developing culturally sensitive means of recruiting and retaining patients in studies must also be addressed. Involvement of the community, physicians, and social service organizations in encouraging participation in clinical trials and adhering to study design -- as well as in participation in riskreduction programs -- is also clearly needed. Fast-track regulatory review will be needed as the global epidemic of AIDS cries out for unusual procedures. Informed consent procedures, including consideration of possible future exclusions from participation in other clinical vaccine trials, will need to be addressed. While the financing of these vaccine trials may be readily accommodated within the current grant funding structure, provision of treatment for those who acquire HIV infection while participating in these studies must be established before the trials begin. Once an effective vaccine has been established, the means and mechanisms for distributing it worldwide and the provisions for monitoring its effectiveness and side effects will need to be worked out. In addition, a means for evaluating new and potentially more effective vaccines within the context of an existing approved vaccine will have to be considered. Clearly, the challenges of HIV vaccine development and vaccine implementation are great and extend beyond the scientific and medical communities. While the AIDS Vaccine 2001 meeting focused primarily on the science of HIV vaccine development and evaluation, we must be cognizant of other key issues as we move forward in this field.
REFERENCES
The following images related to this document are available:Photo images[hs01026t1.jpg] |
| |||||||||

{kind=link}