 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
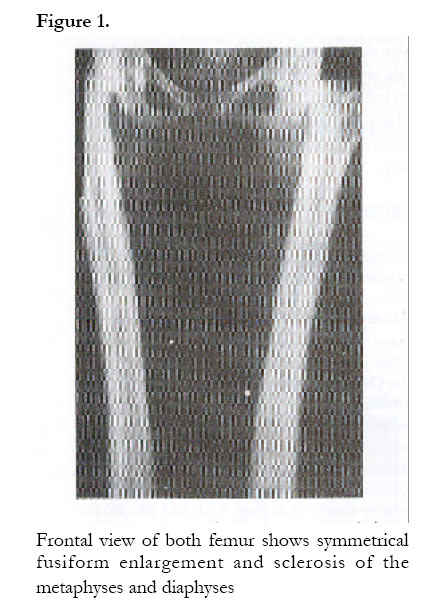
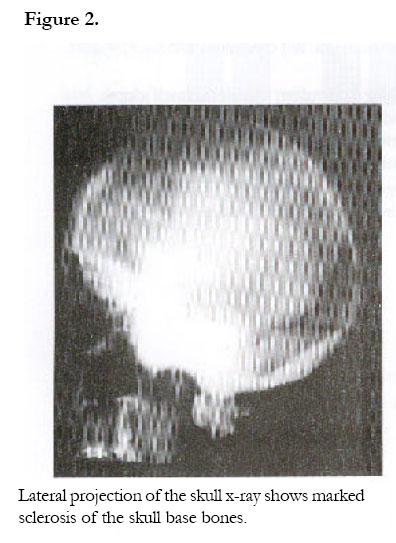
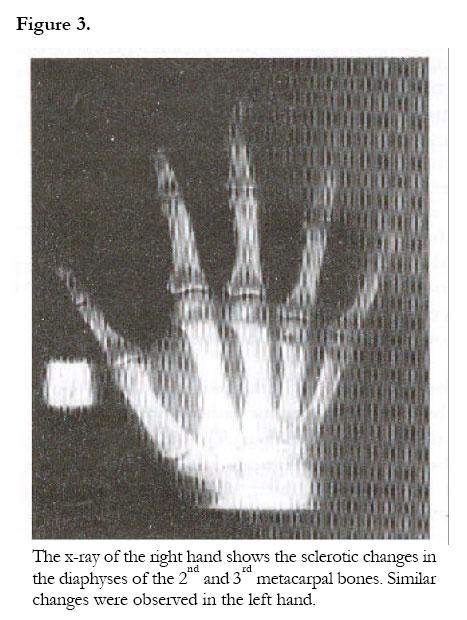
African Health Sciences, Vol. 2, No. 3, December, 2002, pp. 118-120 Case Reports Camurati-Engelmann’s disease: a case report Rosemary Kusaba Byanyima and Jennifer Batuuka Nabawesi Department of Radiology, Faculty of Medicine, P. O. Box 7072 Kampala, Uganda Correspondence: Rosemary Kusaba Byanyima Mulago Hospital Department of Radiology P.O Box 7051 Kampala- Uganda Tel: 256(41) 530137 Email: r_byanyima@hotmail.com Code Number: hs02051 Abstract Camurati-Engelmann’s disease is a rare condition worldwide. No cases have been documented in Uganda. A 26 year old female presented with a history of grinding pain in the limbs for over 20 years. Strong painkillers would temporally relieve the pain. She had an asthenic stature with generalised reduction in muscle bulk. Plain x-rays revealed the characteristic symmetrical thickening and sclerosis of the diaphyses of the appendicular skeleton and skull base, which is pathognomonic of Camurati-Engelmann’s disease. Involvement of the metaphyses of these long bones as well as the metacarpal bones makes this an unusual case. Case Report A 26 year old female Ugandan presented with a history of pain in all limbs for over 20 years. The pain was grinding in nature and at times piercing. Strong painkillers relieved the pain temporarily. She is the only sibling of her mother who died of postpartum haemorrhage. The antenatal history was unremarkable and delivery was vaginal at term. The developmental milestones were normal. Her mental capacity at school was good and she performed well despite interruptions in attendance due to body aches. Menarche was at 20 years following induction with contraceptive pills. The menses last 4 days and the cycles are 21 days. There was no history of a similar illness in the family and no family history of sickle-cell disease. Her appetite was noted to be good and bowel habits normal. There was no history suggestive of heavy metal poisoning or long-term ingestion of vitamin supplements. At the age of 8 years she underwent radiological investigations at the same Hospital and a diagnosis of Engelmann’s disease was made for which she received oral steroids and analgesics with good response. On physical examination she was of asthenic stature with reduced muscle bulk. She was not anaemic or jaundiced. Systemic examination was unremarkable; in particular there was no tenderness over her whole body and no hepato-splenomegally. Cushingoid facial features due to prolonged steroid therapy were observed. Thyroid function tests were normal. Plan x-ray findings (Fig.1, 2 &3): A skeletal survey showed irregular thickening and sclerosis of the cortex in the diaphyses of the appendicular skeleton with relative sparing of the metaphyses and epiphyses except for the femora and humeral bones. The medullary cavity of these bones was narrowed. In the hands the 2nd and 3rd metacarpals were involved. The pelvic bones around the acetabulum were sclerotic too. The skull x-ray showed marked sclerosis and thickening of the bones of the skull base and inner tables of the frontal and occipital bones. The pituitary fossa and the rest of the cranial vault appeared normal. The bones of the vertebral column were osteoporotic but well aligned. There was no paravertebral shadow. The bone age was normal. The soft tissue bulk was reduced but fat planes were preserved. The radiological features were consistent with Camurati-Engelmann’s disease. There were no features to suggest complications like fractures, osteoarthrosis or malignant change. Other radiological investigations were unremarkable and notable was absence of hepatomegally and splenomegally at ultrasound. Discussion Engelmann’s disease (Synonyms; camurati-Engelmann disease; progressive diaphyseal dysplasia; osteopathica hyperostotica multiplex infantilis) is a rare condition worldwide. By 1994 there were only 170 documented cases worldwide, and no case has ever been reported from Uganda. Failure of bone resorption and remodelling at the sites of intermembranous ossification such as the cortex of tubular bones, the skull base, mandible and the middle segment of clavicle is the abnormality notable in this condition1. The mode of inheritance is autosomal dominant with variable penetrance 1, 2. Radiologically it is recognised by the symmetrical fusiform enlargement and sclerosis of the cortex of long bones due to endosteal and periosteal thickening. This condition primarily involves the diaphyses and spares the metaphyses and epiphysis as demonstrated in this case except for the involvement of the metaphyses of the femora and humeral bones as well as the 2nd and 3rd metacarpal bones, which makes this a unique case. Resultant narrowing of the medullary cavity causes anaemia and secondary hepatosplenomegally, which were not present in this patient 1,2,3,4,5. Amorphous increase in density of the skull base bones may cause impingement on the cranial nerves causing symptoms like perceptive hearing loss and serious otitis media due to narrowing of the auditory canal and the tympanic cavities 6,7. Although there was significant sclerosis of the skull base bones, this patient had no impingement symptoms as yet. The reduced muscle bulk is due to myopathic changes associated with thickening of the blood vessel walls suggesting an autoimmune disorder. This has been supported by the observed elevation of monoclonal antibody carrying monocytes and response to steroid therapy 5. The diagnosis in this case was based on the clinical history, characteristic radiological findings and response to steroid therapy. The differential diagnosis includes: Multiple diaphyseal sclerosis (Ribbing disease) a familial disorder which is generally asymptomatic and exhibits asymmetrical long bone involvement; craniodiaphyseal dysplasia which involves only the skull bones and spares the appendicular skeleton and infantile cortical hyperostosis (Caffey’s disease) which starts in the first year of life, and has associated soft tissue swelling characteristically involving the clavicle, ulna and mandible Hyperostotic lesions of hypervitaminosis are also a differential diagnosis 1,2,3,5,8. Progressive diaphyseal dysplasia may be associated with bone malignancy in a rare condition known as Hardcastle syndrome 9. This is an autosomal recessive skeletal dysplasia characterised by diaphyseal sclerosis, medullary stenosis, pathological fractures, bone infarcts and malignant transformation. Since there was no clinical suspicion of Hardcastle syndrome, the patient was not subjected to Thallium isotope scans for tumour screening.
Copyright © 2002 - Makerere Medical School, Uganda The following images related to this document are available:Photo images[hs02051f2.jpg] [hs02051f1.jpg] [hs02051f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}