 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
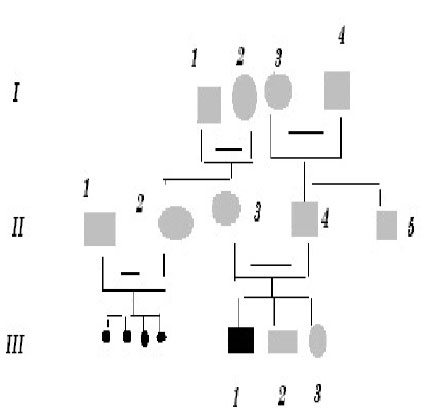
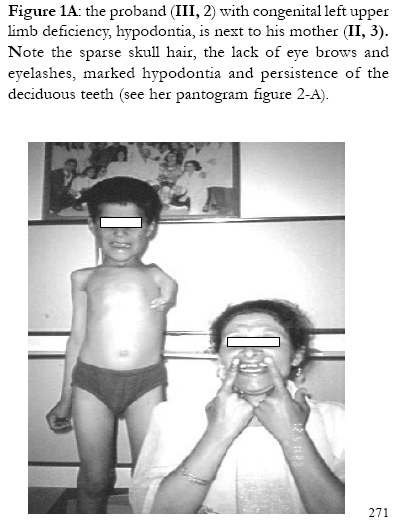
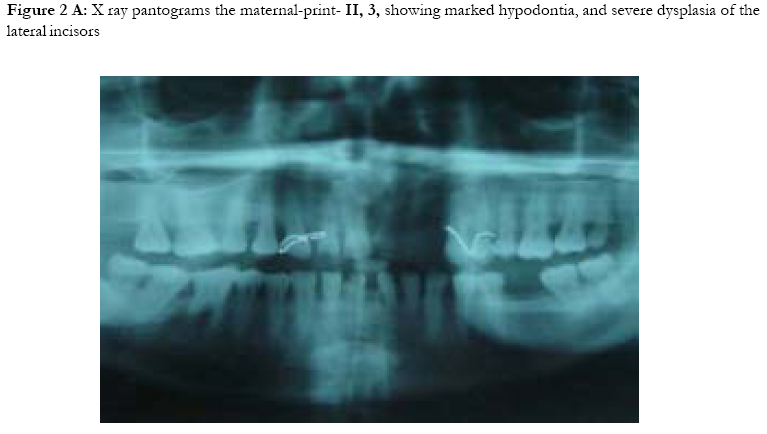
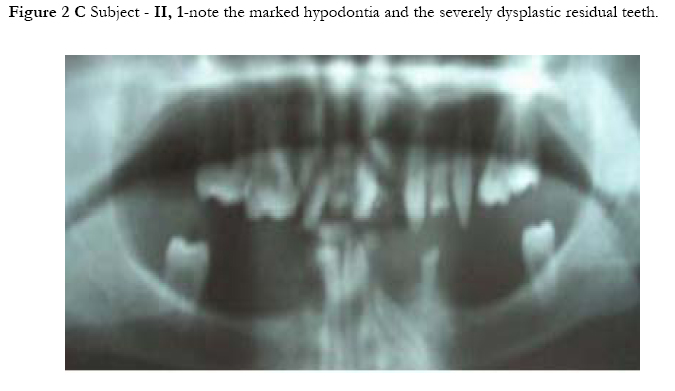
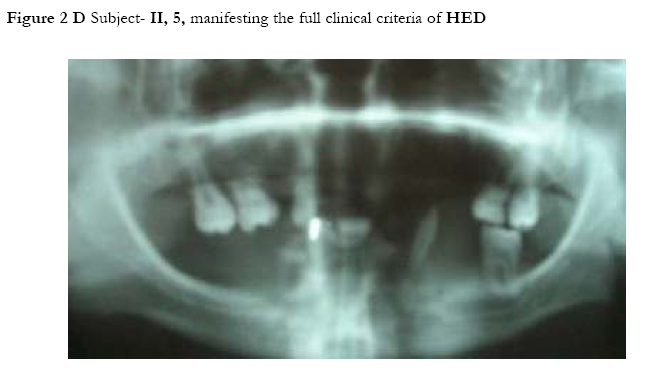
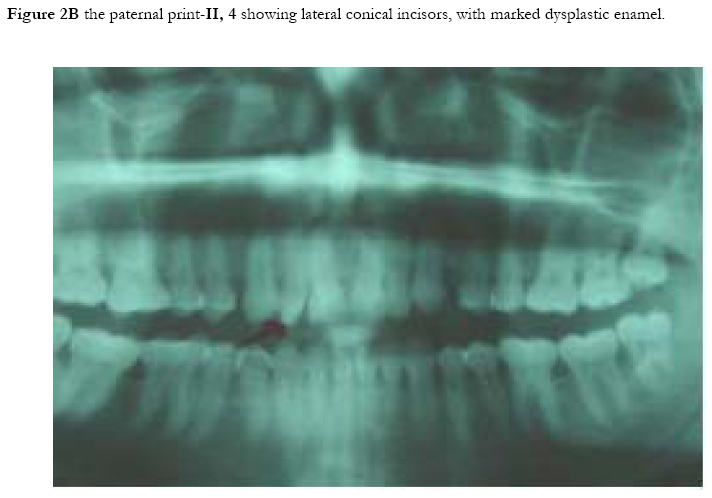
African Health Sciences, Vol. 5, No. 3, September, 2005, pp. 270-275 Subtotal amelia in a child with autosomal recessive hypohidrotic ectodermal dysplasia Ali Al Kaissi1, Farid Ben Chehida2, Nabil Nassib1, Hatem Safi1Mrad Djnziri1, Maher Ben Ghachem1 and Hassan Gharbi2 1 Service d’Orthopedie Infantile-Hopital d’Enfants de Tunis Code Number: hs05046 Abstract: We report an inbred Tunisian family, in which the proband manifested signs of hypohidrotic ectodermal dysplasia, subtotal amelia, scoliosis and left renal agenesis. Two other family members had the full clinical criteria of hypohidrotic ectodermal dysplasia, characterized by deficient sweat glands, hypodontia, hypoplasia of the mucous glands, and fine hair. Nine family subjects had variable clinical expression of the disorder. Key Words: Hypohidrotic ectodermal dysplasia (H E D), subtotal amelia, dysplastic ears IntroductionPatients with hypohidrotic ectodermal dysplasia (HED) manifest sparse scalp hair, eyebrows and eyelashes, and have little body hair. They do not sweat and often present in infancy with high fevers. Hypodontia, abnormally shaped teeth (mostly conical), pigmentation and dryness of the skin around the eyes are additional features. Affected subjects have a prominent forehead, a saddle nose, prominent lips and a hoarse voice. Both lacrimal and salivary secretions can be reduced. Obligate carrier females in the X-linked form of this condition might have sparse hair. We present a family that is clinically not different from the X-linked type, but given the consanguinity and equal severity in males and females, represents the autosomal recessive form of this condition. In addition to the above mentioned cardinal criteria of the disorder, we encountered one member of this family with subtotal amelia, congenital left renal agenesis, scoliosis, and we suggest that maternal hyperthermia during gestation, was an additional factor in the pathogenesis of the disorder. This is the second case report regarding children with congenital limb deficiency born to mothers with hypohidrotic ectodermal dysplasia (Al Kaissi et al,.2002). Case reportThe proband, a 7-year-old male child (III, 2- Figure 1, A) was referred to the department of paediatric orthopaedics because of left sided subtotal amelia. He was the product of an abnormal gestation in that the mother experienced feeble uterine movements, bouts of vaginal bleeding in the first trimester, and intractable bouts of hyperthermia. The latter was a consistent complaint of the mother in all her gestations, and was thought to be secondary to urinary tract infections, but all the investigations ruled out this possibility. The mother (II, 3 ) is a 36 year old gravida 3, mortality one who is married to a 41 year old first degree relative. Her first born child (III 1), died at the age of 17 months at a regional hospital in southern Tunisia because of uncontrolled persistent hyperthermia and dehydration. The proband was born at full term, with a birth weight, length, and head circumference all around the 10th.percentile. He was confined to a baby care unit for ten days because of respiratory distress secondary to pneumothorax, which was treated and he was discharged soon afterwards. His motor developmental was delayed, possibly because of his congenital deficient upper limb, and chronic secretory otitis media. At the age of 4 years a myrinigotomy was performed, and because of his conductive hearing loss he had difficulties at school. He was hyperactive and showed poor concentration. His growth was on the 50th. percentile. There was marked frontal bossing, relatively depressed nasal bridge, sparse scalp hair, sparse eye brows, severe hypodontia, dysplastic ears, and soft skin. He had a congenital upper left limb deficiency with of a trace of the humerus (only six cm), connected to a rudimentary humeral bone which had split off in the prenatal period from another humeral part, which is barely connected to the lateral upper border of the scapula. There was no trace of a shoulder joint, and because of this the lateral head of the clavicle was without any connection. There was a duplication of dysplastic radial bones with abbreviated ulna, all with free mobility because of the lack of any articular attachment. Neither the elbow joint, nor inferior radio-ulnar joints were present .The radial bones connected to severely abbreviated 4th and the 5th fingers only. A juvenile type of thoraco-lumbar scoliosis had developed because of the absence of the normal structural skeletal balance. A congenital agenesis of the left kidney was found. Chromosomal studies on the child and his parents, revealed normal results. Family historyThe parents are first degree relatives and come from a highly inbred family. The third youngest female sib (III, 3) is a 5- years- old with normal phenotype but with very fine soft, hairless skin, sparse eye brows, protruding, dysplastic ears, and sparse scalp hair. The child was frequently absent from school, because of bouts of bronchial asthma, with allergic, eczematous skin. Her cousin II,3, was similar (note her pantogram –figure 2,A, and her spouse’s pantogram, B), He himself is short has very sparse hair and smooth fine skin. The mother and her maternal female sib (II 2), who is a 26-year-old, had bouts of status asthmaticus, and is now receiving corticosteroids, regularly. She has a cushinoid face, and truncal obesity. She has had multiple spontaneous abortions (four in a row) followed by secondary sterility. She is married to a first degree relative II, 1, who manifests the full-blown picture of hypohidrotic ectodermal dysplasia (note his pantogram-figure 2 C, which was taken at age of 18 years). Interestingly, subject II, 5, is the uncle of the proband, a-22-year-old man, who shows the full clinical criteria of the disorder. Note his pantogram- figure 2 D, the severe hypodontia and the residual, conical, lateral incisors. Family Tree
Shadowed squares and circles are variably affected family subject with Hypohidrtotis ectodermal dysplasia, the small black dots (generation) are multiple spontaneous abortions, whereas blackened III, I is juvenile death. Discussion Hypohidrotic ectodermal dysplasia is characterized by, hypodontia, hypotrichosis, and hypohidrosis. Congenital bone deficiency can occur in ectodermal dysplasia, especially association with ectrodactyly and clefting (the E.E.C syndrome) but that syndrome (Bixler, 1972) is inherited in a autosomal dominant fashion and neither clefting nor ectrodactyly were part of the clinical spectrum in our family. In our family the eleven subjects were examined and one juvenile death (III, 1) occurred. The phenotype was variable and we encountered only two subjects with the full clinical criteria of H.E.D, whereas the rest of those affected had variable expression of the disorder. Wallis (1988) reported five individuals from a four generation Mauritian family with ectodermal dysplasia and ectrodactyly, but not associated with subtotal amelia as shown in our family. Al Kaissi et al (2002) in a similar study, reported 19 family members with an ectodermal dysplasia, in whom ectrodactyly occurred as an isolated manifestation in one, and tibial aplasia in two other family members. None had facial clefts, but dysplastic ears were part of the syndrome. No member had subtotal amelia. Pierri et al (2000) reported a male patient with bilateral upper limb amelia , facial clefts, and bilateral renal hypoplasia, but no mention was made of an ectodermal dysplasia.. Caroline et al (2002) reported a sporadic case of micrognathia-limb reduction anomaly, with total amelia, but again, this was not part of an ectodermal dysplasia. In our patient with H.E.D we encountered skeletal (subtotal amelia), and visceral (unilateral kidney), anomalies and in addition there were dysplastic ears, eczema and bronchial asthma. The latter might have been due to deficient bronchial mucous glands. Hyperthermia is an important issue in this family, and it might be that the multiple spontaneous abortions in the maternal female sib (II, 2) who is married to another first degree related man (II, 1) are related to anomalies caused by hyperthermia. This at present is speculation. Hyperthermia was a consistent feature, in all of the proband’s mother’s pregnancies but vigorous investigations have shown nothing else of significance. A number of animal studies has shown severe maternal hyperthermia during the first one third to one half of gestation, to be teratogenic, Edwards (1971), showed that maternal hyperthermia can have adverse effects on growth, development and brain function., but studies on humans are limited in this field. Most families with HED are X-linked and recessive inheritance is much rarer. In our family the consanguineous marriages and the equal severity in both sexes of the classic form of hypohidrotic ectodermal dysplasia, raises the possibility of autosomal recessive mode of inheritance, Passarge et al., (1966).. It is possible that severe hyperthermia in the first and early second trimesters can cause fetal disruption. The temperature must usually be higher than 39 degrees centigrade for more than a day.. Characteristic abnormalities are neuronal heterotopias, microcephaly, neural tube defects, seizures, mental retardation, microphthalmia, micrognathia, mid-face hypoplasia, abnormal external ears and cleft lip and palate. Hirschsprung’s disease (Lipson, 1988) and Moebius syndrome (Graham et al., 1988) are other possible associations. The aim of this study was to indicate the possible association of the genetically determined disorder, hypohidrotic ectodermal dysplasia, with abnormal foetal development due to maternal hyperthermia AcknowledgmentWe wish to thank Professor Michael Baraitser (Institute of Child Health-Clinical and Molecular Genetics-University College London), for his help in reviewing this paper. References
Copyright © 2005 - Makerere Medical School, Uganda The following images related to this document are available:Photo images[hs05046f2a.jpg] [hs05046f2d.jpg] [hs05046f2b.jpg] [hs05046f2c.jpg] [hs05046f1b.jpg] [hs05046f1c.jpg] [hs05046f1a.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}