 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
African Health Sciences, Vol. 6, No. 1, March, 2005, pp. 51-54 Fetal hemoglobin during infancy and in sickle cell adults Dominic Edoh*, Charles Antwi- Bosaiko, Dominic Amuzu Zoology Department, University of Ghana, Box 67, Legon, Accra, Ghana Code Number: hs06011 Abstract Background: Fetal hemoglobin has been implicated in the modulation of sickle cell crisis though it is functional during infancy. Introduction
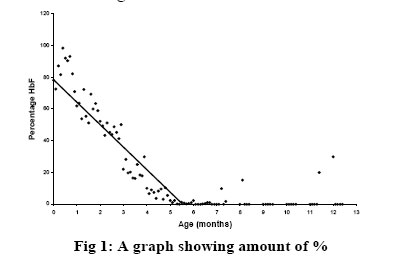
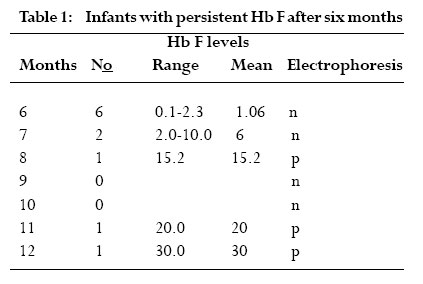
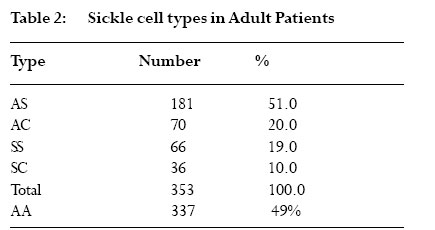
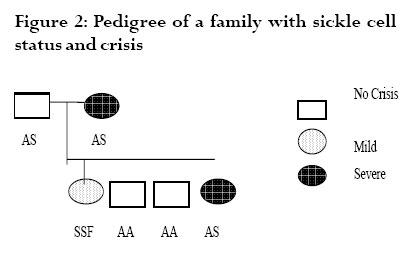
The red blood cells function mainly to transport gases into and out of cells. This is facilitated by a structural component of hemoglobin, which has the ability to bind with gases. Three types of hemoglobin are synthesized in humans depending on the stage of development. Embryonic hemoglobin is produced before birth, fetal hemoglobin (HbF) during foetal life, and adult hemoglobin after birth. Foetal hemoglobin production is switched off soon after birth although the time of switch over is not know. Hemoglobin is structurally made up of 2 alpha and 2 non-alpha chains; in addition, HbF has 2 – gamma chains and the predominant adult blood HbA1 has 2 beta chains. A replacement of glutamic acid of the beta chain by valine at the 6th position gives rise to a sickle cell disorder. This change, called hemoglobin S (HbS), is an abnormal hemoglobin 1 . On exposure to low oxygen concentration, the HbS precipitates into elongated crystals appearing as sickled, instead of a biconcave disc. Sickle cell disease is characterized by occlusion events in the vascular that results in pain, organ failure and, occasional, death 2,3,4,5 . Studies have revealed that HbF usually disappears from red blood of infants after about 6 months 6 . However the exact time of disappearance of HbF may vary and the signal that determines the switch from fetal to adult hemoglobin is not known. Very small quantities of HbF, < 2%, have been detected in the blood of some adults. HbF may be found in certain conditions such as childhood anaemia, myeliod leukeamia, hereditary persistence and sickle cell crisis 7,8 . It is not clear how HbF concentration will affect the severity of crisis in sickle cell individuals and in sickle cell variants. The objective of this study is to determine the waning time of HbF during infancy and HbF persistence in later life. In addition to determine the sickle cell variants of patients attending a sickle cell clinic and to investigate the possible function of HbF among various members of a family with sickle cell variants. Materials and methods The cross sectional study was purely laboratory based with partial clinical observation. Children on admission, either routinely after birth or for simple illness such as diarrhoea and malaria were recruited. Those with transfusion and or complicated diseases were excluded. Sampling Venous blood was collected from 100 infants, aged 0- 12 months, on admission at a military and children’s hospitals in Accra. About 2 ml blood was drained into EDTA sequestering tube and washed 3 times with saline solution. The blood in tube was centrifuge and frozen for 1 hour to thaw and hemolyse. Carbon tetracycline (CCl4) was added to separate cell membrane from the stroma. Blood collected from adults at the clinic was also hemolysed and stored for later use. Electrophoresis and colorimetric methods The electrophoresis technique allows separation of hemoglobin types normal (A), fetal (F) and sickle (S, C) on a cellulose acetate membrane strip in the order A>F>S>C. About 20µl lysate was applied in a 2 cm line one third way from the cathode of a membrane strip.The membrane was placed in an electrophoresis tank containing Tris buffer and 200 volts applied for 40 mins. The strip was removed with a forceps, hot oven dried and analyzed. Colorimetric method with alkaline was used to estimate HbF levels because HbF is more resistant to denaturation. About 0.2ml hemolysate was added to NaOH solution for 1 minute and the reaction stopped with (NH4)2SO4.The precipitate was filtered out on paper and the optical density of the filtrate was measured with a spectrophotometer and the %HbF calculated. The %HbF was calculated as the optical density (OD) of foetal hemoglobin divided by OD of standard multiply by 100. Sickle cell type At a sickle cell clinic at Korle bu hospital in Accra, patients who reported with sickle cell crisis and related ailments were recruited and tested for their sickle cell types using the electrophoresis method described above. Those recruited had clinical manifestation of sickle cell disease. A sickle cell positive patient with HbF had her family members recruited and further examined for their sickle cell status.They were recruited because the family members were available since they resided on the University of Ghana campus. Both laboratory based electrophoresis and clinical observations were done on the family members. A structured questionnaire was administered to the family members to determine the form and frequency of crisis they experience. Results HbF levels in infants Out of the 100 infants recruited, 10 were dropped due to difficulties in obtaining enough blood. A total of 90 infants were examined for their fetal hemoglobin, HbF, levels.Ten each were aged 1, 2, 3, 4, 5 and 6 months and five each aged 7, 8, 9, 10, 11 and 12 months.There were about equal numbers of both sexes; females were 52%. HbF levels was highest at birth reaching 98.30% and a mean of 84.60%. It decreased steadily to a mean of 21.65% at 3 months, below 2% at 5 months and wane off by 6 months as shown in fig 1. By electrophoresis 36% (n=32) were positive. However, high levels of HbF were found in 11 infants aged 6 months and above. Six infants aged 6 months, had HbF that ranged from 0.1% to 2.3%. None of these HbF was detected by paper electrophoresis. One of the 7 months old had HbF as high as 10%, which was negative by electrophoresis. One each of the 11 and 12 months infants were positive for HbF by electrophoresis and had higher HbF titres; 20% and 30% respectively (Table 1). All other recruits aged 6 to 12 months were negative for HbF. Sickle cell types at clinic At the Korle-bu sickle cell clinic in Accra, 690 adults, aged 1 to 80 years, were examined for their sickle cell types. Of these 49% (n=337) had normal blood type AA and 51% were sickle celled.Table 2 shows the sickle cell types of the examined group namelyAS,AC, SS and SC.The AS type was predominant with 51% and the least type SC was 10%. All those examined had sickle cell crisis and one patient of SS type who had measurable HbF levels had her family members further examined for their sickle cell status. In another study, 68 sickle cell positive adults by microscopy were examined for HbF presence. Those with SS were 33 (49%), SC were 29 (43%) and SSF 6 (8%). HbF in a sickle cell family A family of six adults, including 2 parents, were examined for their sickle cell types and the presence of HbF.The father and the mother were aged 45 and 34 years and the offspring 20, 17, 14 and 8 years. By paper electrophoresis the parents had blood types AS each and the children SS, AA, AA and AS. Fetal hemoglobin, HbF was present in the blood of the father and the daughter.The mother and the only son with AS had no detectable HbF. HbF was quantitatively higher in the father (AS with 35%F) who experienced no sickle cell crisis, and lower in the daughter (SS with 15%F) who had mild crisis.The mother and the son, both with AS types, experienced severe crisis. The pedigree of the family is shown in Fig 2. Discussion This study showed that HbF was higher, about 98%, at birth and decreased at a rate of about 10% per fortnight till about 6 months when it wane off almost completely. This confirms earlier studies that the waning period of HbF is between 5 and 6 months 6.The waning off of HbF in infants is due to the switching off from fetal to adult hemoglobin. Since the time and the mechanism of the HbF switch off is not known, it was not possible to compare and correlate HbF disappearance by the two methods. Some infants (n = 10) above 6months in this study were found with high HbF of 10% or more. HbF is elevated in certain conditions including aneamia, β-thalassemia or sickle cell crisis 7,8,9. Probably these children with elevated HbF may be suffering from one of these diseases, although the particular one was not investigated. The study confirmed earlier findings that HbF is higher in sickle cell adults sufferers than in non-sickle cell subjects. It also presents primary observation on the relations between HbF levels and severity of crisis in sickle cell types within a family.A father of AS with high HbF had no crisis and the daughter carrying SS with F had mild crisis. The other members with AS types had crisis and were found to have no observable HbF. This suggests that HbF increases in sicklers and most likely to protect them from crisis.We do not know whether the mother and son were both only AS since AS individuals rarely experience severe painful crisis. It may be possible that the nylon membrane electrophoresis method used was too insensitive to separate out other abnormal hemoglobins or else 40 minutes was too short for better separation. Futher studies are needed on this family with more sensitive electrophoresis method and full clinical observation done. Konotey et al. 10 showed that 35% (n=60) of adult sicklers had frequent crisis and their HbF levels were below 20%, and those with mild crisis had HbF levels above 20%. In that study, the mean HbF level for SS patients was 7.7%, control AS patients was 0.83%, and healthy crisis 1.08%. Middle east sicklers who do not have frequent crisis were also found with 30% HbF 10. The HbF level of 35% observed in sickle cell patients in this study is comparable to other studies in Ghana, Middle East and Britain 11,12,13. These values were far higher than those obtained in sickle cell patients in Nigeria.We do not know whether environmental factors affect the level of HbF. Further studies are necessary to establish the relation between HbF levels and severity of crisis in different sickle cell types and in different settings. Till this date correction of hemoglobin disorders such as sickle cell disease is not available at the DNA level. Conventional treatment is aimed at reducing the patient’s complains and preventing chronic or irreparable organ damage.Transmission therapy has a place in major hemoglobin disorders including sickle cell diseases. In sickle cell hemoglobin therapy, the goal is to dilute the HbS containing erythrocytes to ensure better tissue oxygenation and thus prevent vascular occlusion. Increasing the patient’s amount of fetal hemoglobin is the most promising target. Previous studies involving administering the drug hydroxyurea (HU) to Black American sickle cell patients, increased HbF significantly and revealed a significant decrease of the frequency of yearly crisis 14. Research towards the understanding the mechanism of the transition from fetal to adult hemoglobin synthesis and possible reversion to fetal haemoglobin is important. Conclusions This study demonstrated that fetal hemoglobin (HbF) waned off at 6 months after birth, but HbF persist in some adults. Some members of a sickle cell family with HbF had no or mild crisis suggesting HbF is a potential tool for sickle cell disease therapy. Acknowledgments We are indebted to Mr. Francis Gbogbo and Prof. Coker for critically reading the manuscript. References
Copyright © 2006 - Makerere Medical School, Uganda The following images related to this document are available:Photo images[hs06011f1.jpg] [hs06011t2.jpg] [hs06011t1.jpg] [hs06011f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}