 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
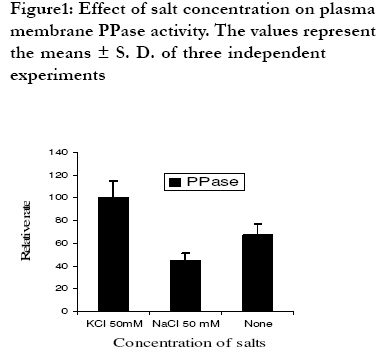
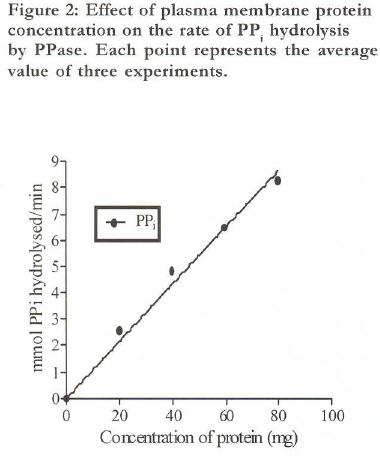
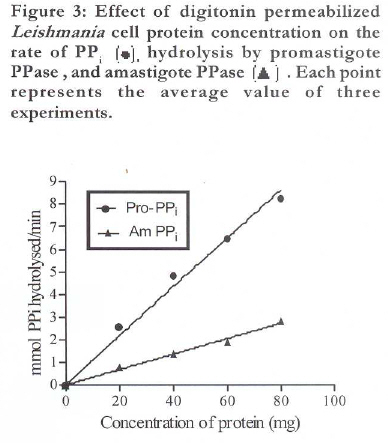
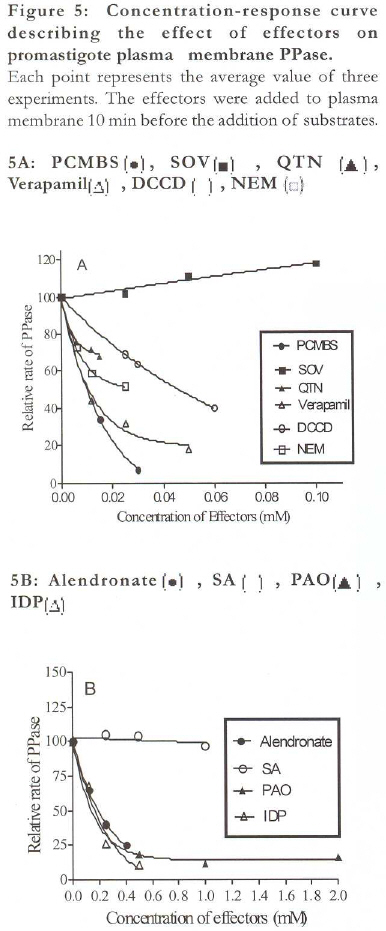
African Health Sciences, Vol. 9, No. 4, December, 2009, pp. 212-217 Original Articles Characterization of plasma membrane bound inorganic pyrophosphatase from Leishmania donovani promastigotes and amastigotes Sen SS1, Bhuyan NR2, *Bera T 1 1. Division of Medicinal Biochemistry, Department of Pharmaceutical Technology, India Code Number: hn09055 Abstract Background: Currently, a major problem in the management of visceral leishmaniasis or kala-azar, especially in the
Indian subcontinent, is the growing unresponsiveness to conventional antimonial therapy. Membrane bound
pyrophophatase (PPases) do not exist in plasma membrane from mammals. Thus,
H+-PPases from Leishmania plasma membrane
might be potential target in rational chemotherapy of the disease caused by Leishmania parasites. Keywords: Leishmania donovani; pyrophosphatase; plasma membrane Abbreviations used: PPase- Pyrophosphatase, NEM- N-ethylmaliemide, PAO- Phenylarsineoxide, KF- Potassium fluoride, DCCD- N,N'-dicclohexylcarbodiimide, PCMBS-Parachloromer curibenzenesulfonate, SA- Sodium azide, SOV- Sodium orthovanadate, IDP-Imidodiphosphate, QTN- Quercetin, LDC- Leishmania donovani cell, DPC- Digitonin permeabilized cell, PM- Plasma membrane, PPi Pyrophosphate, gp- Glycoprotein, Pro- Promastigote, Am- Amastigote Introduction Protozoa parasites are responsible for important diseases that threaten the lives of nearly one-quarter of the human population world-wide. Among them, leishmaniasis has become the a leading cause of death, mainly due to the emergence of parasite resistance to conventional drugs1. Leishmania donovani, the causative agent of visceral leishmaniasis, encounters a wide range of pH values in its life cycle. The gut of the phlebotamine insect vector is extremely alkaline, whereas promastigotes of L. donovani invade host cells via acidic lysosomes2. Mechanisms to cope with this varied environmental pH and maintain cytosolic pH homeostasis might involve the use of proton pumps (H+-ATPases and H+-PPi ases) on both plasma membrane and internal membranes. These transport proteins specifically and actively mobilize ions, generating chemical gradients across a membrane. This movement of ions is vital for numerous cellular functions ranging from energy production, motility, nutrient uptake, ionic homeostasis, intracellular signaling, and differentiation, to name a few. Membrane-bound proton-translocating inorganic pyrophosphatases (H+-PPase; EC 3.6.1.1) belong to a new category of proton pumps, distinct from F-, P-, and V-ATPases, which utilize pyrophosphate hydrolysis as the driving force for H+ movement across the biological membranes3. The membrane-bound, proton-pumping inorganic pyrophosphatase was first described4 in chromatophores from the photosynthetic bacterium Rhodospirillum rubrum. Under physiological conditions this enzyme can both synthesize and hydrolyze PPi . It was also demonstrated5,6 that PPi could drive a number of energy requiring reactions in chromatophores including ATP synthesis in the dark7. H+-PPi synthase is the only known alternative to the well-known ATP synthease in biological electron transport phosphorylation8. H+-PPase gene have been reported to occur in Leishmania parasites9. Eukaryotic cells also possess soluble inorganic pyrophosphatases. Pyrophosphate (PPi) is formed as a byproduct in several metabolic reactions, for example, DNA and RNA synthesis. It has to be hydrolyzed in order not to stop these reactions. This is a major function of the soluble, cytoplasmic PPases. Membrane bound PPases do not exist in plasma membrane from mammals10, thus, H+-PPases from Leishmania plasma membrane might be potential target in relation to rational chemotherapy of the disease caused by Leishmania parasites11. In the present work, we demonstrate that L. donovani promastigote and amastigote plasma membrane possesses PPase activity with features in common with the trypanosomatid and plant enzymes4. Our results also indicate that PPi analogues inhibit the PPase of Leishmania parasite. Methods Culture methods12, 13, 14 L. donovani promastigote strain MHOM/IN/1978/UR6, a clinical isolate from a confirmed kala-azar patient , was grown at 24oC on blood agar medium, pH 7.5. The cells were washed at 500x g twice in cold Tris-sucrose-salt solution ( 250 mM sucrose, 50 mM NaCl, 20 mM KCl, 1 mM ethylenediamine tetra acetic acid, 20 mM Tris, pH 7.2) and kept it at 4oC until use. Viability of harvested cells was monitored microscopically by trypan blue exclusion. Amastigote culture method15, 16, 17 L. donovani amastigote strain MHOM/IN/1978/UR6 was grown and maintained as described by Debrabant A et al. Axenically grown amastigotes of L. donovani are maintained at 37 ºC with 5% CO2 by weekly subpassages in MAA/20 (medium for axenically grown amastigotes) at pH 5.5 in 177 cm2 Petridishes. Under these conditions, promastigotes differentiated to amastigotes within 120 hr. Cultures were maintained by 1:3 dilution once a week. The axenic amastigotes remained stable in culture for a long time. Axenic amastigotes were routinely recycled every 10 weeks by differentiation back to promastigotes, and in parallel, initiating a new line of promastigotes. Transformation of amastigotes to promastigotes was performed by centrifugation of amastigotes (1,000×g at room temperature for 10 min), suspension in promastigote medium, and incubation at 25ºC. Under these conditions amastigotes differentiate to promastigotes within 72 hr. Assay of pyrophosphatase activity18 Pyrophosphatase activity, in terms of Pi release, was assayed according to the method of Katewa and Katyare. The reaction mixture contains buffer B [Sucrose 300mM, KCl 50mM, Tris 50mM, pH 7, EGTA 2mM pH 7], 0.5 mM Magnesium acetate, 0.25 mM pyrophosphate disodium together with 25 µg L. donovani plasma membrane preparation in a final volume of 1 ml. The mixture was incubated at 25oC for 15 min, and the reaction was terminated by adding 0.14 ml of 2.5 M HClO4. In the blank experiments, the reaction mixture was incubated, and the protein solution was added after reaction termination. The amount of enzyme catalyzing hydrolysis of 1 µmol of pyrophosphate per minute is taken to the unity. Activity was calibrated with a phosphate standard solution. For enzyme inhibition study, inhibitor was added to the reaction mixture containing enzyme but lacking pyrophosphate. The mixture was preincubated for 10 min at 25oC before starting the reaction with pyrophosphate. Activity values shown below represent means ±S.E. of three independent experiment. Plasma membrane preparation19 Plasma membrane from L. donovani promastigotes was prepared according to the method as described before. Parasites were harvested at a concentration of 108 cells/ml (5000 x g, 10 min, 4°C) washed twice in PBS, and pooled with 125I cells. All subsequent steps were performed on ice or in refrigerated centrifuges. The cell pellet was suspended to 2 x 107 cells/ml in PBS buffer, pH 7.2 that contained 10 mM MgCl2 and rapidly mixed with equal volume of 1 mg/ml concanavalin A in the same buffer. Cell aggregation was apparent within 1 min. After 5 min, cells were gently spined at 1,000 x g for 1 min to remove excess concanavalin A. The supernate was discarded, and the cell pellet was re-suspended in 12 ml of 10mM Tris-HCl buffer, pH 7.5, that contained 10µg leupeptin/ml and 1mM MgCl2. After swelling for 10min in that hypotonic buffer, cells were homogenized by 18-20 strokes in tight fitting Dounce-type homogenizer. Cell lyses and formation of membrane sheets were verified by phase-contrast microscopy. The homogenate was layered over a two- step gradient consisting of 8 ml of 0.5 M mannitol over 4 ml 0.58 M sucrose, both in Tris buffer, and spin at 1000 x g for 20 min. For analysis, material remaining at the top of the 0.5 M mannitol was saved. Large crude plasma membrane fragments were separated as a tight pellet at the bottom of the gradient. This pellet was re-suspended in Tris buffer that contained 1M á-methylmannoside and left on ice for 40 min with occasional mixing. This plasma membrane, free from bulk concanavalin A, were diluted into three volumes of Tris buffer and homogenized by 80 strokes with a glass Douncetype homogenizer. This second homogenate was layered on a single-step gradient that consisted of 20% sucrose in Tris buffer and spun for 30 min at 500 x g. Scrolls and large plasma membrane sheets above the 20% (w/v) sucrose layer was collected by centrifugation at 40,000 x g for 1 hr. The pellet containing the enriched plasma membranes, was re-suspended in Tris buffer. All samples were either assayed immediately or frozen at -20°C for further use. Preparation of digitonin permeabilized Leishmania cell L. donovani promastigote and/or amastigote cells were collected, washed once by buffer A (140mM NaCl, 20mM KCl, 20mM Tris, 1 mM EDTA, pH 7.5) and resuspended in isolation buffer (20 mM MOPS, pH 7.0; 0.3% bovine serum albumin; 350 mM sucrose; 20 mM potassium acetate; 5 mM magnesium acetate and 1 mM EGTA). Cells were permeabilized in separate tube with 50µg digitonin/mg protein and incubated on ice for 10 min. After incubation, the cells were centrifuged at 6,000×g for 7 min. Pellets were re-suspended in assay buffer. Protein estimation20, 21 LDC protein was determined by the biuret method in the presence of 0.2% deoxycolate. 1 mg of promastigote protein corresponds to 1.75 × 108 cells and 1 mg of amastigote protein corresponds to 1.14 × 108 cells. Cytosolic, mitochondrial and the plasma membrane protein were determined by modified method of Lowry et al. Materials All the biochemicals unless otherwise mentioned were from Sigma-Aldrich (St. Louis,MO,USA). Panmede was purchased from Paines and Byrne (Greenford, Middlesex, UK). Statistical analysis All experiments were performed in triplicate, with similar results obtained in at least three experiments. Statistical significance was determined by student's t test. Significance was considered as P < 0.05. ResultsIsolation of plasma membrane Development of a method based on cell disruption by osmotic swelling in presence of a plasma membrane specific lectin concanvalin A and differential centrifugation has proved to be ideal for obtaining a plasma membrane fraction from L. donovani promastigote19. This membrane fraction consists of open membrane sheets with microtubules attached to the membranes22. The utility of the method was assessed by assaying marker enzymes. The activity of tartarate-resistant acid phosphatase, a plasma membrane marker of L. donovani23 was enriched in this fraction. An enrichment of 28 fold was obtained when cell surface plasma membrane was labeled with 125. PPase activity in plasma membrane Activity of PPase was optimum at pH 6.8 (data not shown). KCl greatly stimulated PPase activity. Replacing 50 mM KCl with 50 mM NaCl or 50 mM choline chloride in the buffer resulted in substantial loss of PPase activity as shown in figure.1. Table 1 show that activity of PPase in plasma membrane is located at endoplasmic face of plasma membrane. Permeabilized L. donovani cell showed substantial PPase activity, whereas nonpermeabilized cell did not. However, very little ecto-PPase activity have been observed at exoplasmic face of the plasma membrane. Figure 2 shows PPase activity and the amount of plasma membrane proteins added was linear. Linearity of PPase activity was also observed for digitonin permeabilized cell. It is also evident from Figure 3 that in digitonin permeabilized amastigote cell, PPase activity was lesser than promastigote. Specific inhibitors and stimulators of PPase activity PPi hydrolysis in the plasma membrane and digitonin permeabilized L. donovani promastigote cell was inhibited, in a dose-dependent manner by KF as indicated in figure 4. PPi hydrolysis was inhibited by the PPi analogues, IDP and alendronate. Since IDP is a bisphosphonate, we tested other bisphosphonate (alendronate) used clinically in the treatment of bone resorbtion disorders24 for its inhibitory effect on PPi hydrolysis. The sulphydryl group inhibitors p-chloromercuribenzensulphonate (PCMBS) and phenylarsineoxide (PAO) showed potent inhibition on PPase activity as indicated in figure 5A and Figure 5B. The chemical modification with the trivalent arsenical reagent PAO indicates the involvement in PPase activity of a viscenal sulfhydryl group25. Some compounds used to inhibit H+ATPase activity, such as N-ethylmalimide (NEM) and DCCD26, are also able to inhibit the plasma membrane PPase. F1-ATPase and F1Fo ATPase inhibitor sodium azide27 did not inhibited the PPase activity. Discussion In the present study, we have identified and characterized PPase activity in the plasma membrane of L. donovani promastigote. This is the first report demonstrating biochemically the presence of a PPase in the plasma membrane of L. donovani promastigote. Our results suggest that K+ was necessary for PPase activity, but substantial amount of K+ and Na+ independent PPase was also present in the plasma membrane. Since IDP is a bisphosphonate, we tested other bisphosphonate (alendronate) used clinically in the treatment of bone resorbtion disorders24 for its inhibitory effect on PPi hydrolysis.The chemical modification with the trivalent arsenical reagent PAO indicates the involvement in PPase activity of a viscenal sulfhydryl group25. The significance of the presence of a PPase in the amastigote plasma membrane may be proton pumping to maintain neutral cytosolic pH 31 in an environment of acidic pH of phagolysosome32. Data provided evidence that "pH formation in promastigote and amastigote was partially inhibited by H+-ATPase inhibitor DCCD. This fact suggested that mechanisms in addition to that inhibited by DCCD may also be involved in controlling homeostasis of pHi 31. It appears from our observations that PPase may also play an important role in the extrusion of proton from the cytosol. Conclusion In conclusion, this is the first report of a PPase in plasma membrane of an organism different from plants. Since membrane bound PPases are apparently absent from vertebrates, analysis of the role of PPase in pH regulatory mechanisms of L. donovani amastigote might serve as potential target for putative therapeutic intervention. Acknowledgements Support for this work was partially supported by grants from Department of Science and Technology, Indian Council of Medical Research, New Delhi and R.D. Birla Smarak Kosh, Mumbai, India References
Copyright © 2009 - Makerere Medical School, Uganda The following images related to this document are available:Photo images[hs09055f2.jpg] [hs09055f5.jpg] [hs09055f3.jpg] [hs09055t1.jpg] [hs09055f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}