 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
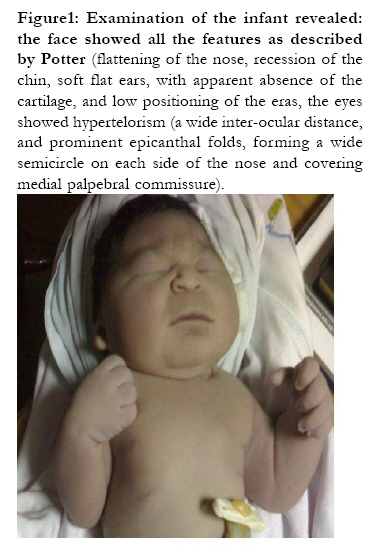
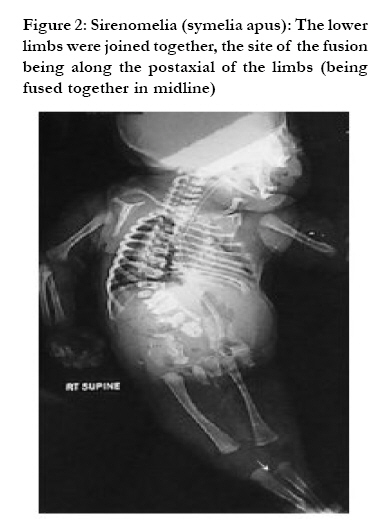
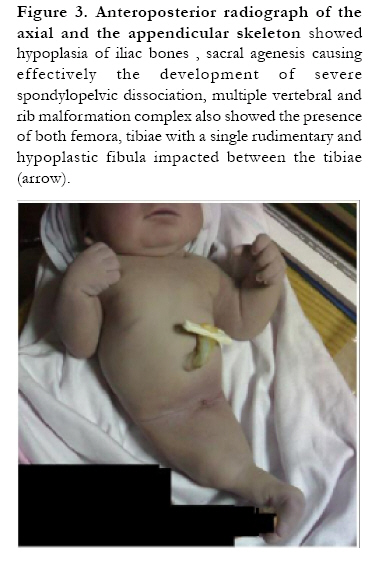
African Health Sciences, Vol. 10, No. 4, December, 2010, pp. 353-361 Case Reports Sirenomelia (symelia apus) with Potter's syndrome in connection with gestational diabetes mellitus: a case report and literature review Al-Haggar M1, Yahia S1, Abdel-Hadi D1, Grill F2, *Al Kaissi A2,3 1Pediatrics Department, Genetic unit Mansoura University
Children's Hospital, Mansoura, Egypt *Correspondence author: Ali Al Kaissi, Ludwig Boltzmann Institute of Osteology, Hanusch Hospital of WGKK and AUVA Trauma Centre Meidling, 4th Medical Department, Hanusch Hospital, Heinrich Collin Strasse, 30 A- Vienna, 1140, Austria, E-mail: ali.alkaissi@osteologie.at Code Number: hs10072 Abstract We report one case of a fetus of sirenomelia sequence with Potters syndrome which showed oligohydramnios and symelia apus. The infant showed absent urinary tract and external genitalia, the legs were fused by skin and had separate bones associated with Potter's syndrome. The mother had a history of gestational diabetes mellitus. Key words: Sirenomelia; Potter's syndrome; Symelia apus; Gestational diabetes mellitus Introduction Sirenomelia, or the mermaid syndrome, is the most extreme example of the caudal regression syndrome. It invariably presents with lower limb fusion, sacral and pelvic bony anomalies, absent external genitalia, anal imperforation, and renal agenesis or dysgenesis. Because of the resultant oligohydramnios, these infants most often have Potter's facies and pulmonary hypoplasia. Prognosis is very poor because of the condition involves variable major anomalies, including bilateral renal agenesis, sacral agenesis and imperforate anus. Only four cases of a surviving infant with sirenomelia have been reported1. Potter syndrome is usually fatal in the first few days of the patient's life; most often, the cause is pulmonary failure. Neonates with the potter sequence have an increased morbidity rate because of respiratory failure, pneumothorax, and acute renal failure during the neonatal period. Infants with Potter's facies have a flattened nose, receded chin, prominent epicanthal folds, and low-set abnormal ears. The case described in this report shows the association of the abnormality with the syndrome described by Potter of renal agenesis, hypoplasia of the lungs, and a typical facies1,2.The condition occurs predominantly in males, with a sex ratio of 2.7: 13. The sex of our foetus is a female. Most cases of caudal regression are sporadic or associated with gestational/maternal diabetes. The condition is thought to be part of a spectrum including imperforate anus, sacral agenesis and sirenomelia4. Gestational diabetes mellitus is not a disease per se, but an abnormal plasma glucose level. Controversies still exist regarding gestational diabetes mellitus (GDM) and its prevalence, screening, clinical management and impact on maternal and infant outcomes5-9. Clinical report A 42-week, 2450 gram term infant was born by a caesarean section to a primagravida mother 24- year- old- with a history of gestational diabetes mellitus. No family history of similar conditions (congenital anomalies) or consanguinity. Prenatal ultrasound showed marked oligohydramnios which was associated with marked restriction of the movement of lower limbs. Later on, just before labour, sonar showed absent bladder and kidneys, marked scoliosis to left side due to extensive vertebral malsegmentation. Elective caesarean section had been undertaken to avoid postmaturity. Examination of the infant revealed; the face showed all the features as described by Potter (flattening of the nose, recession of the chin, soft flat ears, with apparent absence of the cartilage, and low positioning of the eras, the eyes showed hypertelorism (a wide inter-ocular distance, and prominent epicanthal folds, forming a wide semicircle on each side of the nose and covering medial palpebral commissure) (fig 1). The pelvis was of small size and no external genitalia could be seen. The lower limbs were joined together, the site of the fusion being along the postaxial of the limbs (being fused together in midline) (fig 2). Cardiac examination was uneventful apart from ejection systolic murmur maximum on the second left interspace, echocardiographic evaluation revealed no structural anomaly but only haemodynamic abnormalities of persistent foetal circulation secondary to bilateral pulmonary hypoplasia. Radiologically there is hypoplasia of iliac bones , sacral agenesis causing effectively the development of severe spondylopelvic dissociation, multiple vertebral and rib malformation complex also showed the presence of both femora, and both tibiae with a single rudimentary and hypoplastic fibula impacted between the tibiae (arrow) (fig3). Apgar scores were 4, 7 at one and 5 minutes; patient was immediately transferred to neonatal intensive care unit and subjected to assisted ventilation. Postnatal ultrasound revealed absence of the bladder, ureters, and both kidneys. In the subsequent days patients developed deterioration of the ventilatory parameters; chest x-rays revealed air leak. Family refused autopsy studies. Cytogenetic analysis revealed a normal female fetus (46, XX). Discussion A malformation is a primary error of normal development or morphogenesis of an organ or tissue. All malformations are thus congenital or present at birth, although they may not be diagnosed until later, specially if microscopic or if internal organs are involved malformations may be single or multiple and may be of minor or major clinical significance. About 14% of newborns have a single minor malformation, 3% of newborns have a single major malformation and 0.7% of new born have multiple major malformations. The frequency of major malformations is even higher at conception (10-15%), but the majority of these fetuses are spontaneously aborted. a disruption (or secondary malformation) is a morphological defect due to breakdown of a previously normal organ or tissue. Disruptions can arise at any stage of gestation after initial morphogenesis. Similarly, deformations arise after the embryonic period and are alterations in shape caused by unusual mechanical forces. About 2% of newborns are affected, with multiple deformations in one-third. Malformations and deformations coexist, and there is an increased risk of deformation (8%) in the presence of a major congenital malformation, especially of the central nervous system or urinary tract. multifactorial inheritance is the commonest identifiable cause of congenital malformations followed by monogenic and chromosomal abnormalities. Thus, genetic disorders account for at least one-third of all congenital malformations of known aetiology .Deformations are caused by any factor which restricts the mobility of the foetus such as oligohydramnios and so causes prolonged compression in an abnormal posture10. Infants of diabetic mothers have two to three times the average incidence of congenital anomalies. These include neural tube defects, cardiac defects (transposition of the great vessels, coarctation of the aorta, VSD, ASD, cardiomyopathy), situs inversus, renal anomalies (hydronephrosis, renal agenesis, multicystic dysplastic kidney, duplication of the renal tracts), intestinal atresia, and forms of caudal regression including sacral agenesis7,8. Lynch and Wright9 reported a case where the mother had diabetes and the infant had sirenomelia with renal agenesis and an absent right radius. There was also a hypertrophic cardiomyopathy and a bicuspid pulmonary valve. Sirenomelia is thought to occur in about 1 in 65.000 live births and may be more common in one of monozygotic twins. It bears resemblance to the mermaid Greek mythology .Affected individuals exhibit a variable range of defects, including hypoplasia and or fusion of lower limbs, vertebral abnormalities, renal agenesis, imperforate anus, and anomalies of the genital organs11. Previous reports described the extent of the malformation complex in association with sirenomelia. McCoy et al, 12 reported an unusual case with two normal kidneys. Perez-Aytes et al, 13 reported an unusual case where there appeared to be partial fusion of the lower limbs associated with absent feet, but other features of caudal regression and a single aberrant umbilical artery. Akbiyik et al., 14 reported male twins, one of which had sirenomelia and the other just an imperforate anus. Unfortunately zygosity was not established. Shonubi et al, 15 reported sirenomelia in 1 of 2 monochorionic twins. Duncan et al., 16 reviewed 445 cases ascertained for sacrococcygeal malformations and foundthat 12% had sirenomelia, 27% VATER association and 34% what they called the sacrococcygeal dysgenesis association. It was felt that these cases were aetiologically distinct from VATER association and sirenomelia cases. They had fewer anomalies per patient, a lower incidence of anatomical urinary tract anomalies (but a higher chance of neurogenic urinary problems), and a high incidence of CNS anomalies such as spina bifida and hydrocephalus. 28% of cases were born to diabetic mothers. Occasional cases have holoprosencephaly or cyclops17,18 or craniorachischisis19 . Selig et al., 20 reported a family where three siblings and a maternal aunt had renal anomalies consistent with renal dysplasia (renal agenesis, cystic dysplastic kidneys, and urethral valves). A fourth sib had sirenomelia. Both parents had normal renal ultrasound examinations. Managoli et al., 21 reported a case with in addition good evidence of amniotic band disruption (midline disruption and adhesion of the lips by amniotic strings, a disrupted mandible, a bifid tongue, amputated fingers and toes, hands attached to each other and to the chest wall in the midline at the sternum, feet that were attached to the symphysis in the midline by bands, body wall defects), and some other anomalies on examination and at PM (microphthalmia in one eye and megalocornea in the other, genitourinary hypoplasia, anal atresia, bicornuate uterus, no ovaries, hypoplastic lungs, an accessory spleen and an hypoplastic aorta. The sirenomelia (the legs were fused by skin and had separate bones) was in the fetal position. Guidera et al22 reported a case of orthopaedic abnormalities and pelvic dysplasia, fused lower extremities, and osseous fusion of the calcanei, but with normal femurs, tibias, and fibulas. This patient also had significant cardiac and renal abnormalities. Stevenson et al. 23 reported in 1986 that the aetiology of sirenomelia was explained by vascular steal theory. They documented the common feature of a single large artery arising from high in the abdominal cavity, which they thought diverted nutrients from the caudal end of the embryo in a vascular `steal' phenomenon. The associated anomalies are variable and involved multiple organ systems. The most common anomalies include a single umbilical artery, malformation of urinary tract, lower gastrointestinal tract and external genitalia24. In summary Sirenomelia sequence is classified into three groups according to the number of feet present2. The most common of the three conditions is symelia apus, in which both legs are merged completely into a single lower extremity. In this condition both feet are absent or rudimentary. Symelia unipus shows a presentation of one foot, two femora, tibiae and fibulae. In symelia dipus, two distinct feet are present but are malrotated and resemble fins. Our case was compatible with symelia apus associated with Potter's syndrome. Careful diabetic control in the pre-conceptional period and the first eight weeks of pregnancy may lower the chances of congenital anomaly. Sells et al., 25 found that intellectual function in infants where mothers had been well controlled was not different from controls, whereas there was a correlation between delayed development and poor maternal diabetic control, or major congenital malformations in the infant. References
Copyright 2010 - African Health Sciences The following images related to this document are available:Photo images[hs10072f3.jpg] [hs10072f1.jpg] [hs10072f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}