 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
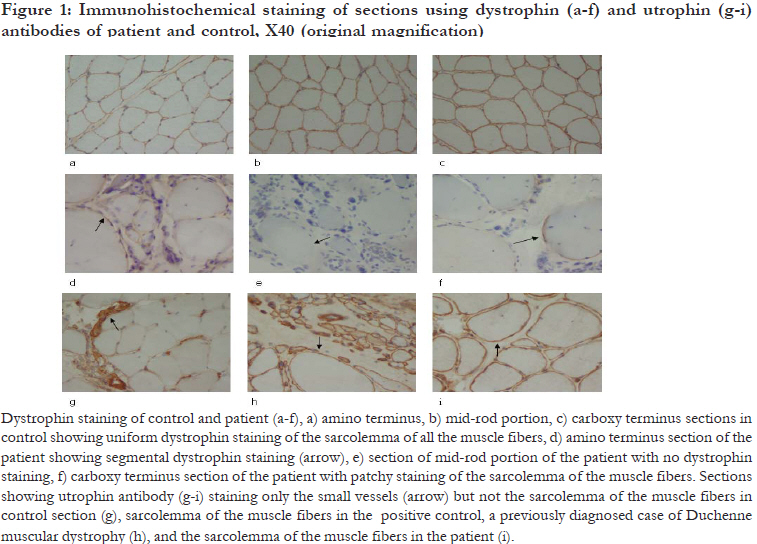
African Health Sciences, Vol. 11, No. 4, Dec, 2011, pp. 607-609 Phenotype-Genotype analysis of dystrophinopathy caused by duplication mutation in duplication mutation in Dystrophin gene in an African patient Peddareddygari LR1, Pillai BH1, Nochlin D2, Sharer LR3, *Grewal RP4 1 Neurogenetics Foundation, Cranbury, New Jersey, USA. Code Number: hs11120 Abstract Background: The dystrophinopathies, duchenne muscular dystrophy (DMD) and Becker muscular dystrophy are common X-linked genetic myopathies resulting from mutations in the dystrophin gene. Duplication is an uncommon mechanism of mutation occurring in about 5% of DMD cases. The global prevalence of DMD is reported as 1/18,000 males. There is little clinical or epidemiological data on African patients. Keywords: Duchenne muscular dystrophy, exon 8 and 9 duplication, genotype-phenotype analysis. African Health Introduction Dystrophinopathies, Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD), is are common X-linked disorder caused by mutations in the dystrophin gene and clinically characterized by the childhood onset of progressive weakness. The world-wide incidence of DMD is 1/3500 male births with an overall prevalence of 1/18,000 males, while the incidence of BMD is approximately 5 / 100,000.1, 2 There are relatively few investigations in Africans. In studies performed of indigenous South African patients, the overall prevalence of BMD was reported to be 1/ 755,000 and 1/ 100,000 for DMD. A prevalence of DMD as low as 1/ 250,000 was reported in the indigenous black population. In this population, the most frequent mutations are deletions that have been detected at a frequency ranging from 26 % to 63%.3,4 It is possible that lack of facilities and expertise could be responsible, at least in part, for the paucity of clinical and epidemiological data on the dystrophinopathies in Africans. The dystrophin gene is approximately 2.3 Mb and contains 79 exons encoding a 14 Kb mRNA that is translated into a 427 kDa protein, dystrophin, containing 3685 amino acids.5 Dystrophin is not only a structural protein but is also believed to have a physiological role in the organization of postsynaptic membrane.6 Mutations in this gene result in DMD, BMD or outliers who have a clinical phenotype in between that of DMD and BMD and X-linked dilated cardiomyopathy. The majority of the mutations reported in this gene are deletions and point mutations and only about 5% involve duplications.5, 7 These duplications occur most frequently near the 5' end of the gene and in 22% of cases involve exons 6 and 7. In general, the frequency of the duplications decreases to 1% as the 3' end of dystrophin gene is approached.8 The exon 8 and 9 duplication mutation has been previously reported in patients of Asian/ Caucasian descent. However, the phenotype has not been described. We present genotype-phenotype analysis of an African patient of Ghanaian descent with dystrophinopathy caused by duplications of exons 8 through 9 of the dystrophin gene. Case report We evaluated a nine-year old boy who, the parents noted, was having increasing difficulty climbing stairs and running. The onset of these symptoms was gradual and started at least two years prior to evaluation. His past medical history was unremarkable for birth, delivery and the development of major motor milestones. However, over the last two years, he stopped participating in sports and was having increasing difficulty running. He also started to have difficulty arising from a sitting position. The remainder of the general medical review of systems was unremarkable with no cardiopulmonary complaints. General medical examination was unremarkable. A neurological examination revealed a normal mental status with normal cranial nerve and sensory examinations. Cerebellar testing was normal. He had reduced stretch reflexes in his arms and unobtainable reflexes in his legs with flexor plantar response. Muscle strength testing showed mild proximal muscle weakness (MRC grade 4/5) in his arms and legs with a Gower sign when arising from the floor. There was no significant pseudo-hypertrophy of his calves. Examination of his gait revealed exaggerated lordosis. Family history revealed a one year old sister who was normal. Both children are the product of a non-consanguineous marriage of parents who came from Ghana. There are six paternal and five maternal siblings and no other family member, including the grandparents, on either side was affected with any neuromuscular disorder. Routine serum chemistries were normal except the following: CPK9625 U/L (normal value: 24-204 U/L), AST 127 IU/L (normal value: 0-60 IU/ L) and ALT 206 IU/L (normal value: 0-55 IU/L). A cardiac evaluation revealed no abnormalities. Given the history and clinical examination, the diagnosis of Duchenne muscular dystrophy was strongly suspected. Immunohistochemical staining of muscle biopsy sections of the vastus lateralis employing dystrophin and utrophin antibodies showed extensive replacement of muscle by collagen and adipose tissue. Fiber phagocytosis was present without inflammation. The mid rod portion of dystrophin failed to stain in all muscle fibers. However, in some of the fibers, antibodies to the amino and carboxy terminals did stain indicating the presence of protein expression. Overall, the biopsy was consistent with a dystrophinopathy [Figure - 1]. In addition to the muscle biopsy, genetic testing was carried out by a commercial laboratory using Southern blot and densitometric analysis of Hind III digested genomic DNA. The DNA was obtained from peripheral blood white cells of the patient and his mother and probed with dystrophin cDNA. It demonstrated a duplication of exons 8 through 9 of the dystrophin gene in both the patient and his mother. The presence of a duplication of exons 8 and 9 was supported by an alternative technique, the Multiplex Ligation dependent Probe Amplification (MLPA) analysis. Discussion This patient has the clinical features of dystrophinopathy confirmed by both genetic testing and muscle biopsy. Given the lack of family history and the high spontaneous mutation rate of this gene, it is likely that a sporadic mutation occurred in the maternal lineage. The exon 8 and 9 duplication has been identified in twenty eight cases worldwide [ Belgium (2), Canada (2), USA (6), UK (1), Japan (1), Denmark (1), Australia (1), Estonia (2), Hungary (3), Taiwan (1), France (7) and Italy (1) ], listed in the Leiden Muscular Dystrophy pages [http:// www.dmd.nl/ ]. Twenty one of these were diagnosed with DMD and seven with BMD/DMD. However in these cases no phenotype data is provided and there is no correlation with muscle biopsy. It is interesting that the muscle biopsy from this patient shows evidence of residual dystrophin staining excluding the mid portion of the protein [Figure - 1: d, e and f). Whether there is residual functional activity is not known, but we do note that he does have a slightly later age of onset than typical cases of DMD. However utrophin, which is normally restricted to the neuromuscular junction is over expressed in DMD and is present throughout the sarcolemma as seen in this patient [Figure - 1: g, h and i). In addition, our patient has no cardiac involvement which is unusual since a high percentage of DMD patients show cardiac involvement by age six years.9 The disease process is not mild and the patient is too young to specifically diagnose him as BMD. Although patchy dystrophin staining is observed in the carboxy and amino terminals, no dystrophin is found in the mid-rod portion. This labeling pattern leads us to presume that this patient is an outlier presenting with clinical phenotype in between DMD and BMD. Our report extends the epidemiology of exons 8 through 9 duplication in the dystrophin gene causing a dystrophinopathy to include individuals of diverse African populations and is the first report in a patient of Ghanaian descent. It also confirms the vulnerability of this gene to recurrent mutations at this site. Acknowledgements We would like to acknowledge Dr. G.K.Dhaliwal for editorial assistance. References
Copyright © 2011 - African Health Sciences The following images related to this document are available:Photo images[hs11120f1.jpg] |
| |||||||||

{kind=link}