 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
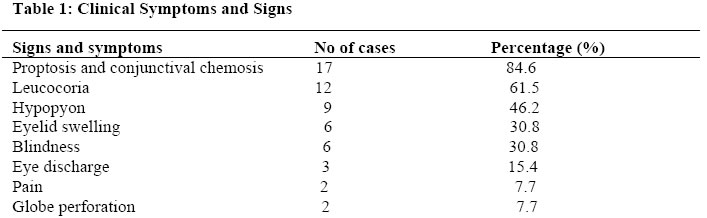
African Journal of Health Sciences, Vol. 13, No. 1-2, Jan-June, 2006, pp. 117-123 RESEARCH ARTICLE Retinoblastoma - a clinico - pathological study in Ilorin, Nigeria* Joshua F. A. Owoeye1*, Enoch A. O. Afolayan2, Dupe S. Ademola-Popoola1, 1. Departments of Ophthalmology; 2.Department of Morbid Anatomy and Histopathology,
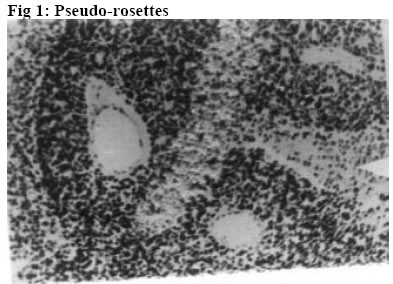
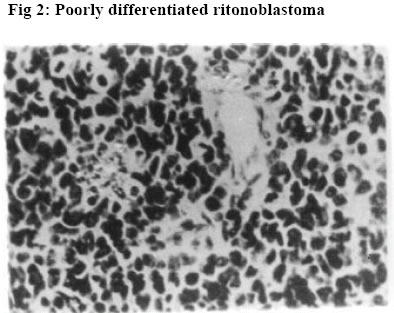
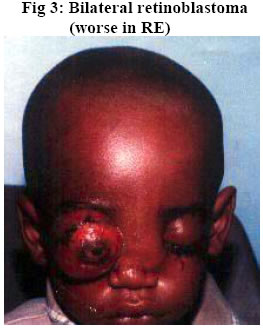
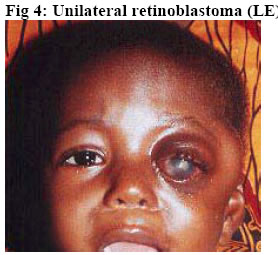
University of Ilorin Teaching Hospital, Ilorin. Kwara – State. Nigeria Code Number: jh06016 SUMMARY Retinoblastoma is the commonest childhood primary malignant intraocular neoplasm that is often characterized by spontaneous regression. They display photoreceptor differentiation. This study provides the clinical presentations and histological profiles of retinoblastoma in Ilorin, Kwara – State, in the North Central geo-political zone of Nigeria. A retrospective study of clinically and histologically verified retinoblastoma at the University of Ilorin Teaching Hospital, Ilorin, Kwara – State, Nigeria from January 1989 to December 2000 was undertaken. The clinical and histological features were analyzed using the patient’s case folder and surgical pathology records. There were 20 patients, 9 males and 11 females (M:F ratio 1: 1.2), age range from 5½ months to 6 years with 23 eyeball tumours histologically confirmed retinoblastoma during the study period. Proptosis with chemosis was the most common clinical presentation (84.6%). Bilaterality was 15% in this study. Enucleation and Exenteration combined with chemotherapy were offered to 15 (75%) and 5 (25%) patients respectively. A poorly differentiated type with extensive areas of tumour necrosis was the commonest histological pattern. Thirteen (65%) of the patients died before completing the course of chemotherapy.[Afr J Health Sci. 2006; 13: 117-123] *Published online before print Introduction Retinoblastoma (RB) is a rare embryonic tumour but commonest primary malignant intraocular tumour of childhood [1, 2]. In the United States of America, retinoblastoma occurred in 1 of every 15000 live births3, whereas in developing countries of Africa and Asia reports showed that it occurred in 1:18000 live births [4, 5, 6]. Retinoblastoma accounted for more than 50% of all ocular and orbital malignancies of all age groups in African series [5, 6, 7, 8]. However in Caucasians it is the second most common tumour following malignant melanoma [3, 9]. Retinoblastoma occurs in sporadic (60%)– usually unilateral or germline [hereditary] (40%) forms; the later manifesting either as unilateral or bilateral disease and characterized by early onset and predisposition to developing secondary non-ocular, intracranial malignancies termed Trilateral Retinoblastoma [10, 11]. Most cases of trilateral retinoblastoma, which occurs in about 8% of heritable retinoblastoma [11, 12, 13] is found in the pineal region, but it can also occur in the suprasellar and parasellar regions [12, 13]. The RB gene is located in the long arm of chromosome 13 (13q14) and the gene is believed to be a recessive suppressor; thus, it requires the loss, deletion, mutation or inactivation of both copies of the gene at the 13q14 locus for RB to develop which correlates with the two “hits” theorized by Knudson [14]. Prenatal and postnatal prediction of susceptibility to inherited RB using recombinant DNA markers is now possible [15]. This would make genetic counseling for familial (hereditary) RB more accurate, leading to an earlier tumour detection and effective therapy. The clinical presentation of RB includes leucocoria, strabismus, conjunctival chemosis, proptosis, and even blindness, which are attributed to late presentation and diagnosis [2, 16]. In spite of the treatment modalities such as enucleation, radiotherapy, photocoagulation, chemotherapy and even exenteration as in extra-ocular involvement, the mortality from RB in our environment is very high [2, 5, 6, 16]. Various studies on RB have been carried out in some centers of tertiary health institutions in Nigeria [5, 6, 17, 18]. The department of ophthalmology in UITH was established in 1988 and during the period under review had staff strength of 2 Consultant Ophthalmologists and 4 trainee ophthalmologists (resident doctors). The hospital is a tertiary institution and serves as a referral center for some states (with a combined population of about 14 million people) in the North Central geopolitical zone of Nigeria. The hospital is however not exactly equidistant to these adjoining states. Kwarastate has a population of about 2.3million with Ilorin, the state capital, (where UITH is sited) having a population of about 600,000. The hospital lacks facility for radiotherapy and often depends on such facility in another teaching hospital about 180kilometers away. The present communication is therefore designed to illustrate the clinical and histological features of RB as seen in our center and would be the first communication in that regard from this center to the best of our knowledge. Materials and Methods A total of 23 ocular surgical specimens from 20 patients with clinical diagnosis of RB at the ophthalmology department of UITH, Ilorin, Nigeria, from January 1989 to December 2000 and registered at the pathology department for histological verification were retrospectively studied. The age, sex, presenting symptoms, duration of symptoms, site, management and other adjunct treatment were obtained from the patient’s case notes and analyzed. Some case notes of patients whose parents refused admission and treatment were excluded from the study. Dilated funduscopy under general anaesthesia with binocular indirect ophthalmoscope was done for the patients for the detection and or exclusion of RB in the other eye. Paraffin sections stained by standard haematoxylin and eosin (H&E) were retrieved and reviewed by one of the Authors. Special stains, Methyl green pyronnin (MGP) for calcium deposition and pearl blue reaction for haemosiderin pigments were employed where necessary. Each optic nerve was examined for involvement in tumour spread up to the line of transection. One case of Burkitt’s lymphoma from a 15-year-old patient was excluded from the study leaving us with 20 patients in the study. The histological diagnosis was based on cell morphology, degree of differentiation, presence of Flexner-Wintersteiner rosettes, optic nerve infiltration by tumour cells to the cut end of the optic nerve and tumour necrosis. Clinical photographs of some of the patients as well as photomicrographs were also included for illustration. Results Twenty-three cases of retinoblastoma were diagnosed from 20 patients during the 11 year interval which accounted for 33.3% of the total number of ocular and orbital tumour cases seen at the eye clinic and 10.5% of childhood malignancies during the same period at UITH Ilorin. There were 9 males and 11females, giving a male to female ratio of 1:1.2. The ages of the children ranged from 5½months to 6years with a mean age of 29months (SD 0.78). No single case was reported in an adult, and the only case reported in a child of 15 years was reviewed to be Burkitt’s lymphoma using special staining of Methyl green pyronnin method. There was no significant age difference between the sexes (males mean age of 27months) and (females mean age of 31months). The commonest clinical presentation in our experience was proptosis with associated conjunctival chemosis (84.6%). This was followed by leucocoria 61.5%, and hypopyon 46.2%. Eyelid swelling and blindness were observed in 30.8% of the cases each -Table 1. Other forms of presentation were eye discharge, pain and globe perforation. Lymphadenopathy (due to histologically confirmed tumour metastasis) was observed in pre-auricular and submandibular lymph nodes in 4 patients each, detected in 3 cases. Autopsy was not performed to ascertain intracranial involvement or the extent of tumour metastasis to other sites. Three (15%) patients had bilateral retinoblastoma, while 17(85%) cases were unilateral with 9 (45%) and 8 (40%) cases involving the right and left eye respectively. The 3 patients with bilateral RB were all males with an average age of 15months but 31 months for patients with unilateral retinoblastoma. A positive family history was denied in the bilateral cases; however an unconfirmed case of similar ocular problem among the siblings was reported by one of the parents of the unilateral cases. Enucleation was done for 17 (73.9%) of the patients while 6 (26%) eyes had exenteration. All the patients received first course of chemotherapy regimen of Vincristine (IV 2mg / m2 weekly x 12 doses, IV Cyclophosphamide 2.5mg / kg body weight every other week and IV Actinomycin D 0.0015mg / kg /day for 1- 5days, then at 2 weekly interval x 6 doses. Table 2 shows the histological features. Nineteen (82.6%) of the 23 cases were poorly differentiated. This is characterized by diffuse sheet of round and polygonal retinoblasts with hyperchromatic nuclei with scanty thin rim cytoplasm, mitotic figures, delicate vascularized stroma, and tumour necrosis. The remaining 4 cases were moderately differentiated type characterized by occasional Flexner- Wintersteiner rosettes. None of the cases in our series was well differentiated. Microscopic infiltration of the optic nerve alone by malignant cells was identified in 16 (69.6%) cases. Other histological or degenerative findings were pseudo-rosettes (Figures 1 and 2), necrosis (97.0%), dystrophic calcification (39.1%), basophilia (17.4%), haemosiderin pigments and cholesterol slits. Eleven (55%) of our patients including the 3 bilateral cases died while on admission. The rest (9 patients) were lost to follow up after the 1st course of chemotherapy and presumed dead based on their clinical condition while on admission with signs of metastasis Discussion In this study, RB accounted for 33% of all cases of ocular and orbital malignancies in our center that compares well with the findings in other studies where the incidence varies from 34 – 55 percent [6, 19, 20]. Similarly, our present experience of RB accounting for 10.5% of all childhood cancer and the commonest childhood intraocular tumour corroborates findings in other African series [19, 20, 22] but high when compared with developed countries where 2.4 – 4% has been reported [2, 23]. We reported a male to female sex ratio of 1:1.2. This agrees with the observation of no sex predilection in other studies [14, 17, 23, 24] but differs from Olurin et al where approximate ratio of 2:3 was observed [17]. Our patients appeared to present very late as the average age of the children at the time of diagnosis was 29 months Though this is comparable to similar findings in the African series [6, 8, 17, 19], it is high when compared with the reports from the western countries [14, 23, 24]. However the average age (15 months) of the 3 bilateral RB was found to be lower than that of unilateral cases that agrees with other studies [16, 17]. Gallie et al [25] attributed the peak age frequency of retinoblastoma occurring soon after birth to the susceptibility of developing retinal cells to carcinogenic factors. Thus retinoblastomas are rarely diagnosed after the age of 5 years in both sexes, as by that time there are no target cells for tumorgenesis and retina differentiation is complete. The late presentation in our study could be due to among others, lack of awareness of the disease, delayed referral prompted by consultation with traditional medical practitioners and poor affordability and accessibility to available medical facilities. The afore mentioned factors as well as the relative recent establishment of the department of ophthalmology in this hospital could also be responsible for the seemilingly low number of cases recorded in this study as compared to the one reported by Akang et al [17] done in the premier teaching hospital in the country. There might be a need for a comparative study in the future of the incidence and prevalence of RB in this environment. Figures 3 and 4 show the typical pattern of presentation in our environment. No single case of RB was reported in an adult in this series which agrees with the findings of Olurin et al [19]. Proptosis with conjunctival chemosis was the commonest clinical presentation manifesting in 84.6% of our patients. This is at variance with findings in other studies where leucocoria and strabismus were observed [16, 17, 23]. This picture is attributed to delay in presentation of the patients for diagnosis and treatment. This opinion is supported by the claim that proptosis is the result of orbital involvement [16]. Also Erwenne et al concluded in their study of 158 consecutive cases of RB that extra ocular disease was strongly dependent on the age at diagnosis and lateness of referral [25]. The incidence of bilateral RB in this series is 15% that agrees with the findings in some other African series [17, 19]. In Zambia it is 4 % whereas in the western countries such as Germany, England and Wales, the incidence is higher [22, 23]. The disparity may be associated with incomplete data or due to higher incidence of unilateral (sporadic) RB over bilateral cases in Africa. Often, information of diseases in the preceding pedigree is withheld deliberately for socio-cultural reason of stigmatization or due to ignorance as to the relevance of such information or outright lack of knowledge of the cause of death of the members of the family. However, the survival rate of patients with retinoblastoma is low in this country due to high mortality associated with late presentation and poor facility. This translates to less transmissible mutant genes surviving to the reproductive age, hence, a lower incidence of hereditary retinoblastomas in the developing countries. But should such survivors procreate? This needs a thorough genetic counseling in our environment. There is the need for a good health policy in our environment to make health facilities affordable and accessible to the majority of the people. More than half (55%) of our patients had distant tumour metastasis involving pre-auricular (4), submandibular (4) and the scalp (3). This is high compared with the study conducted by Akang et al [17] where metastasis also involved the regional lymph nodes, scalp and the skull. This is associated with late presentation of our patients as none of the bilateral RB cases who presented at an earlier age and onset of disease had visible metastasis. Absence of post mortem examination in all the cases might also be responsible for nondetection of secondaries at the skull and long bones, brain, liver, pancreas and other visceral organs as reported in other studies [1, 16, 17]. The treatment modalities of RB include eyeconserving treatments like transpupillary thermotherapy, brachytherapy, radiotherapy, photocoagulation, cryotherapy and systemic chemotherapy [6, 16]. Enucleation with optic nerve is the treatment of choice for most unilateral RB whereas in cases of early diagnosis of bilateral cases, enucleation of the worse affected eye and conservative treatment of the better eye is the preferred mode of treatment. But in this present communication, our patients presented late and due to lack of radiotherapy facilities in our center, all the 20 patients had either enucleation - 17 patients (85%), or exenteration –3 patients (15%) in combination with chemotherapy. Histologically the two characteristic arrangements of cells that are pathognomonic of RB are Flexner- Wintersteiner rosette and Fleurrette representing an attempt to differentiate into photoreceptor cells. Welldifferentiated tumours are characterized by the presence of Flexner-Wintersteiner rosettes. In this communication, 19 eyes (82.6%) were of poorly differentiated type - Figures 1 and 2. This is high when compared with other studies [17, 23]. In this study too, the mortality rate is high in comparison with other studies [17, 23] as 11 (55%) of the 20 patients died while on admission. This could be due to, among other reasons, late presentation with associated optic nerve invasion and distant metastasis as this has been found to occur usually within 1 year of diagnosis and a child is considered cured if there is no metastatic disease within 5 years of retinoblastoma diagnosis [26], however, with metastasis, death occurs within 6 months [26, 27]. Also optic nerve invasion has been found to be one of the predictive factors for death from retinoblastoma [26, 28] for which reason prophylactic chemotherapy with adjuvant radiotherapy has been advocated in cases of RB with poor prognostic indicators [29]. A multicenter collaborative study in our environment in this respect would be necessary to reduce the mortality from this disease. Also genetic counseling should be an integral part of the treatment of these cases in our environment regardless of the laterality. This however, could be difficult due to lack of facility for such and paucity of information (as in our situation in this study) or where available due to genetic mosaicism [30]. The number of patients that was lost to follow up and presumed dead is also high (9 patients). Tracing these patients by the social work department is usually difficult and often fruitless due to wrong, non-traceable addresses in the case notes. Conclusion Retinoblastoma is the commonest malignant intraocular tumour of childhood, mostly of poorly differentiated type histologically and characterized by high mortality in our center. There is need for improved accessibility to health facilities and the importance of genetic counseling should be emphasized. References
Copyright 2006 - African Forum for Health Sciences The following images related to this document are available:Photo images[jh06016t1.jpg] [jh06016f3.jpg] [jh06016t2.jpg] [jh06016f2.jpg] [jh06016f4.jpg] [jh06016f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}