 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
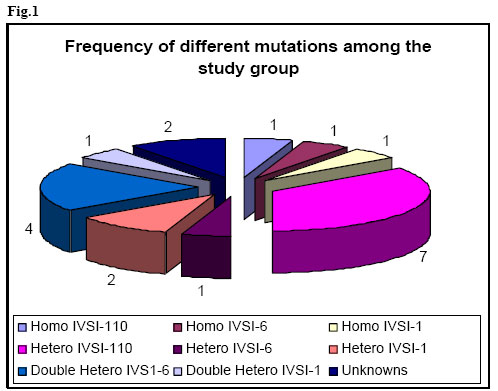
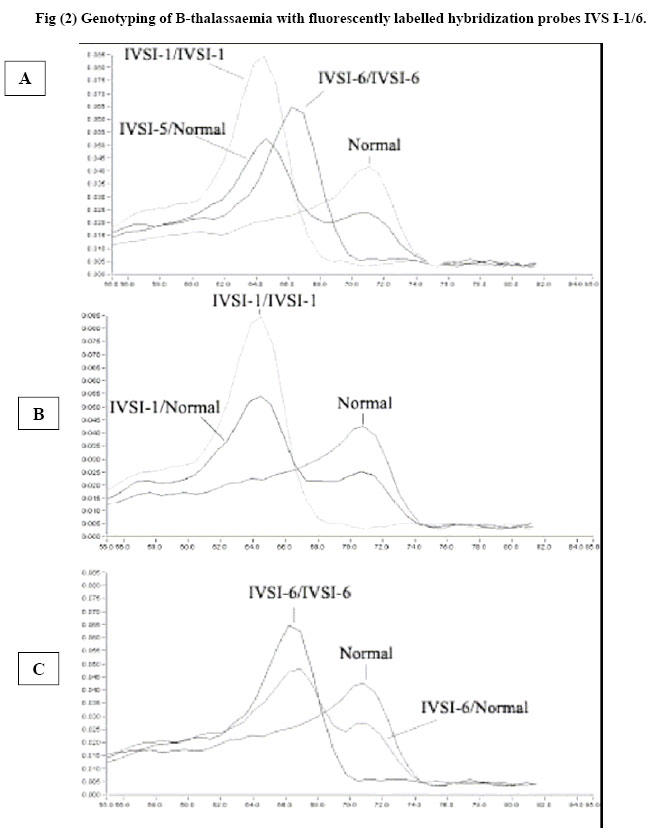
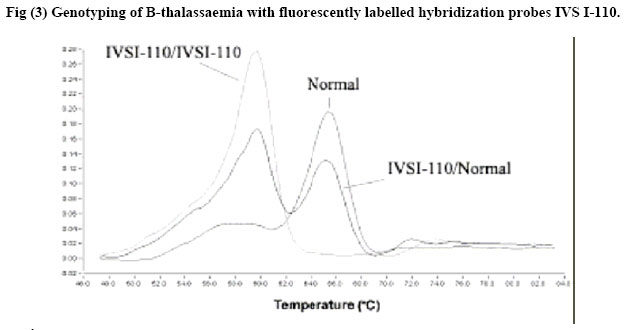
African Journal of Health Sciences, Vol. 13, No. 3-4, July-Dec, 2006, pp. 70-77 Rapid screening of β-Globin gene mutations by Real-Time PCR in Egyptian thalassemic children Taghrid M Gaafar¹, Amal M ELBeshlawy², Mona I Aziz, Heba N Abdelrazik¹ 1. Clinical Pathology Department, 2. Pediatric Department, Faculty of Medicine, Cairo University 14 Aly El Gendy, 6th district Nasr City, Cairo, Egypt Code Number: jh06031 SUMMARY Thalassemia is one of the most common genetic disorders in Egypt. With the total population of 70 million, there are approximately 600,000 affected individuals and more than 20 million thalassemia carriers. Thalassemia is therefore one of the major health problems in Egypt. B-Thalassemias are priority genetic diseases for prevention programs. Rapid genotype characterization is fundamental in the diagnostic laboratory, especially when offering prenatal diagnosis for carrier couples. Introduction of the real time PCR has made a revolution in the time taken for the PCR reactions. We present a method for the diagnosis of the common mutations of the B-thalassemia in Egyptian children & families. The procedure depends on the real-time PCR using specific fluorescently labeled hybridization probes. The melting temperature for each of the specific probes obtained after the PCR reaction permits the identification of the specific mutation. Genotyping of 20 thalassemic children attending the hematology clinic of the children specialized hospital and 10 controls was done using Real-time PCR and the conventional Amplification Refractory Mutation System (ARMS) technique. Analysis revealed identical results to most of the patients and they were further checked by the sequencing results of the DNA samples. The established method is a robust, fast and straight forward assay that allows the detection of the common B-thalassemia mutations in Egypt. The described LightCycler system protocol can rapidly screen for many B-globin gene mutations. Introduction Formerly the distribution of thalassemia had been mainly limited to the areas from the Mediterranean across the Middle East through Southern Asia to Southeast Asia in the so called ‘thalassemia belt [1] However, recent migrations of people have spread thalassemia genes throughout the world. Furthermore, there has been a major transition in the demography of common illnesses over the past years. [2] This change includes a remarkable decline in childhood mortality due to infections and malnutrition in some regions. As a result, many infants with serious genetic disorders such as thalassemia can now survive the early months of life and live long enough to require treatment for their hematologic disorders. Given the demographic population shifts, thalassemia is at present considered to be a global health problem. [2] In spite of the more than 170 different Bthalassaemia mutations described around the world (http://globin.cse.psu.edu/globinlhtml/huismanltha lsl) [3,4], only five or six of them are responsible for more than 80% of the thalassaemic alleles detected in the different geographic areas with a high prevalence of B-thalassaemia. [5] Thus, in the countries bordering the Mediterranean basin, the major mutations of the B-globin gene are CD-39 (C62434T) and IVS 1-110 (G62388A) [6]. The IVS 1-6 (T62284C) mutation is also relatively frequent, and, in Egypt, the mutations IVS I-I (G62279A), CD-37 (G62429A) and CD-6 (-A62206) are also observed [7]. The ability to perform rapid DNA analysis for genotype characterization has become an increasingly important requirement for the clinical diagnostic laboratory. Currently, a wide variety of methods exist for detecting point mutations in a DNA molecule. Classic methods include oligonucleotide hybridization, endonuclease restriction analysis of PCR products, the Amplification Refractory Mutation System (ARMS), denaturing gradient gel electrophoresis, and direct sequencing. All of the above methods require several hours, and sometimes days, for complete diagnosis extensive processing, and some of them use radiolabelled oligonucleotides with potential contamination issues and problems with disposal of radioactive waste [8]. Consequently, there is a need for more rapid, high-throughput assays. Recently, systems have been developed that integrate microvolume rapid-cycle PCR with fluorometry, offering real-time fluorescent monitoring of the amplification reaction for quantitative PCR and/or characterization of PCR products for rapid genotyping without the need for post-PCR sample manipulation [1,2]. The LightCyclerTM (Roche Molecular Biochemicals) is one such system. The detection of potential sequence differences (usually single-nucleotide polymorphisms) for genotyping applications involves the use of two fluorescently labeled probes that hybridize to adjacent internal sequences within target amplified DNA, one of which covers the region expected to contain the mutation(s). Close proximity of annealed probes facilitates fluorescence resonance energy transfer between them. Probes are designed to have different melting temperatures (Tms), such that the probe with the lower Tm lies over the mutation site(s). Monitoring of the emitted fluorescent signals as the temperature increases detects loss of fluorescence as the probe with the lower Tm melts off the template. A single base mismatch under this probe produces a Tm shift of 5–10 °C, allowing easy distinction between wild-type and mutant alleles. The ability to detect base mismatches under the probe with the lower Tm (mutation detection probe) and the use of two, different-colored probes allows more than one mutation to be screened in a single PCR reaction. Because of its low costs, reproducibility, and ease of handling, the assay is potentially suited for the routine clinical laboratory. [9] In this study we aimed to report the standardization of a real-time detection Materials and Methods Provisional diagnosis of thalassemia syndromes was made based on characteristic clinical manifestations and morphologic red cell abnormalties. Definite diagnosis was confirmed by hemoglobin typing based on the electrophoretic or chromatographic separation of Hb from the blood. Iron deficiency anemia was first excluded before making the diagnosis of thalassemia because it may complicate the diagnosis. [10] Subject: The study included genomic DNA from 30 patients; 20 thalassemics attending the hematology clinic of Abulrish hospital and 10 non-thalassemic subjects. These individuals had been previously tested for the major β-thalassemia mutations (IVS I1, IVS I-6, IVS I-110) using the conventional ARMS method [11] and their diagnosis was confirmed by sequencing. Amplification Refractory Mutation System (ARMS): ARMS primers were designed to detect normal and mutant DNA. The nucleotide at 3' end of the mutant primer was complementary to the change of DNA sequence caused by mutation that was being looked for. A set of internal control primers were included in each assay, to demonstrate the efficacy of the PCR. They amplify a region of DNA a short distance from that amplified by the ARMS primers and produce a 861 bp fragment. Oligonucleotide primers to detect the 3 mutations selected for this study were synthesized according to the following sequence I-Allele specific primers: IVS1-110 Mutant 5'ACC AGC AGC CTA AGG GTG GGA AAA TAG ACT IVS1-6 Mutant 5'TCT CCT TAA ACC TGT CTT GTA ACC TTC ATG IVS1-1 Mutant 5'TTA AAC CTG TCT TGT AAC CTT GAT ACG AAT II-Common primer: 5'ACC TCA CCC TGT GGA GCC AC III-Internal control primer: Control A: 5'CAA TGT ATC ATG CCT CTT TGC ACC PCR reaction mix 5 µmol/L of each ARMS primers and Taq enzyme, 3mM Mgcl2, 400 µM of dNTPs and DNA samples were used in a final volume of 25 µl under the following cycling conditions: Initial precycling denaturation hold at 94°C for 5 min. Then 25 cycles of denaturation at 93°C for 1 min, annealing at 65°C for 1 min, extension at 72°C for 15 min. In the last cycle, extension was prolonged for 5 min. The products were then run on 2% agarose gel using gel electrophoresis and visualized on UV transilluminator to detect the presence or absence of the mutant allele. Mutation detection using specific fluorescently labelled hybridisation probes The method was carried out on the LightCycler (Roche Diagnostics, Mannheim, Germany), and comprised real-time PCR amplification of a β-globin gene fragment of 609 bp using the GLO F and GLO R primers together with a pair of hybridisation probes. One of the probes, the anchor probe, was located in the vicinity of the mutation and was labelled in its 5' end with Red 705. An adjacent sensor probe, was placed over the mutation 2 to 5 nucleotides apart from the anchor probe, and was labelled with fluorescein at its 3' end. Design of primers and probes. Primers and probes were all designed and synthesized by TIB MOLBIOL (Berlin, Germany) GLOF: 5'GAAGAGCCAAGGACAGGTAC Real-time PCR protocol PCR was performed in capillary tubes in 10 µl final volume using 1µl of DNA template (50-100 ng), 1x FastStart LightCycler™-DNA Master Hybridization Probes (Roche Molecular Biochemicals), 0.3 µM of each primer and 4 mM MgCl. The hybridisation probes were used at 0.2µM, with the exception of the IVS 1-1/6 sensor probe, which was used at 0.1 µM. The PCR program involved incubation at 94°C for 10 min. Amplification comprised 45 cycles of denaturation at 94°C for 10 seconds, annealing at 55°C for 10 seconds, and elongation at 72°C for 25 seconds. The mutation detection was achieved by melting curve profiles. The program consisted of an initial denaturation at 95°C for 30 seconds, followed by an annealing at 40°C for 30 seconds to permit the hybridisation of the probes. In the case of the probes for IVS I-1/6, the decrease of temperature was gradual with incubations of 25 seconds at 71°C and 66°C (Tm of the anchor and sensor probes, respectively). Then, the temperature was increased at a rate of 0.1°C/second up to 90°C with continuous fluorescence monitoring. The software provided with the equipment (LightCycler 2) gives the temperature of melting (Tm) of the sensor probe. A base pair mismatch between the sensor probe and template causes a decrease in Tm that can be easily detected by the LightCycler [12,13] Results The Light Cycler method was standardized by analyzing DNA from 10 normal controls not suffering from any kind of anemia. The melting curves for the IVS I-1/6 probes, yielded a Tm mean ± standard deviation of 65.96 ±0.41° C while for the IVS I-110 yielded a Tm mean ± standard deviation of 62.67 ±0.32° C for the 10 normal samples. Each PCR reaction included heterozygous and homozygous (when available) samples for the mutation under evaluation, five controls negative for the mutation, and a DNA-free blank to monitor the absence of contamination. In all sets of experiments for each mutation, the melting curves with the detection probe distinguished the wild-type from mutant alleles, allowing assignment of wild-type, heterozygous, or homozygous status to each sample as expected. No false-positive or false-negative results or contamination was observed in any case. The frequency of mutation distribution in the 20 thalassemic patients is illustrated in Fig (1). The most frequently encountered mutated allele was the IVS I110; 7 (35 %) patients were heterozygous for IVS I110 while only one was homozygous. Four (20%) were double heterozygous for IVS I-110 and IVS I-6 while one patient was double heterozygous for IVS I110 and IVS I-1. Two patients were heterozygous for IVS I-1 while one patient only was heterozygous for IVS I-6. The homozygous groups; IVS I-1, IVS I-6 and IVS I-110 constituted of one patient in each group. In two samples, we observed different melting curves which were close to but distinct from the mutant curves of the detection probes; after sequencing, these samples were found to have theIVS I-5 and the frame shift mutations, which are less common in the Egyptian population. All genotype results were confirmed by routine methodologies, including denaturing gradient gel electrophoresis in combination with ARMS PCR and direct sequencing [3,11,12] The IVS I-1 mutation gave a Tm mean ± standard deviation of 59.07 ± 0.45° C, the IVS I-6 mutation gave a Tm mean ± standard deviation of 61.34 ± 0.27° C while IVS I-110 mutation gave Tm mean ± standard deviation of 66.96 ± 0.58° C. Here the design of the sensor probes of the IVS 1-1/6 and IVS I-110 takes the advantage of the fact that different mutations placed along the sequence produce distinct effects on the Tm [12] Thus, the melting curves permitted the distinction of the normal allele from IVS 1-1 and IVS 1-6 mutations separated by five nucleotides apart. The melting curves obtained immediately after the amplification procedure permitted the identification of the Tm peaks corresponding to normal or mutated alleles in each sample (fig 2A) Detection of IVS I-I and IVS 1-6 mutants with IVS I-1/6 probes Normal and IVS I-6 homozygous (HO) subjects show only one peak corresponding to normal (65.96°C) and mutated allele (61.34 °C), respectively, while IVS 1-6 heterozygous (HE) patient shows both normal and mutated peaks (fig2 C). The IVS I -1 HE patient shows a mutated peak 2°C below (59.07 0c) that of the IVS 1-6 mutation (fig2B). Detection of IVS 1-110 mutation The normal subject showed one peak at 62.67 °C. The IVS 1-110 HE patient showed the peak of the normal and the mutated allele (66.96°C). While HO showed one peak of the mutated allele (Fig 3) Thus the Melting curves in an 18 thalassemic patients indicated homozygosity & heterozygosity for a mutation within the B-globin gene regions covered by the mutation detection probes. On the other hand, we noticed that some mutations, including IVSI-5 and the frame shift mutations had different melting curves, which were distinct from the wild type and the mutated type of both probes. In such cases, the mutations were identified by use of ARMS and by direct sequencing [14]. Discussion The B-globin gene is a relatively small gene (2000 bp) located in the short arm of chromosome 11. [15] Although 180 causative mutations have been reported for B-thalassemia syndromes, the spectrum of mutations and their frequencies in most populations usually consist of a limited number of common mutations and a slightly larger number of rare mutation [16]. With the aim of rapid DNA analysis, real-time PCR has become an important tool in both research and routine clinical diagnostics [1, 2, 17, 18] Use of the LightCycler facilitates, and allows simultaneous multiple mutation detection through the flexible design of detection probes. Twenty sequenced DNA samples with Bthalassemia mutations and 10 controls (with normal B-globin gene) were included in the study. Rapid Screening of B-Globin Gene by Real-Time PCR was considered an appropriate model for developing the described protocol. In the selected samples most of the Egyptian common mutations were found represented with different forms; IVS I-110 homozygous and heterozygous, IVS I-1 homozygous and heterozygous, IVS I-6 homozygous and heterozygous together with double heterozygous mutations of IVS I-110 with IVS I-1 and IVS I-6. We designed a duplicate reaction protocol to screen for the multiple B-globin gene mutations, with the use of four probes, two sensor mutation probes located over the mutated areas and two anchor probes located in the vicinity of the mutated area over a normal Bglobin region. Using detection probes complementary to the wild-type sequence allowed the identification of mutations by an abnormal melting temperature of the probe even if the type of mutation is unknown. This potentially allowed the distinction of any allele with a mutation under the length of the probe, minimizing the costs and time required to screen a spectrum of mutations, as is necessary for most populations in which B-thalassemia is common. This approach was greatly facilitated by the close proximity of many B-thalassemia mutations within the B-globin gene [19]. All results were in complete concordance with the genotypes obtained in parallel by routine methodologies; ARMS and sequencing [8]. Normal controls were identified as wild-types, 7 patients were heterozygous for IVS I-110, 2 patients were heterozygotes for IVS I-1 and one for IVS I-6. One patient was homozygous for each of the previously mentioned mutations. Six patients had double heterozygous mutations; 4 for IVS I-110 and IVS I-6 and 2 for the IVS I-110 and IVS I-1. Two patients had Tm that was completely different from the mutations detected previously, one had IVS I-5 (Tm 53°C) and the other was a frame shift mutation (Tm 50.2°C). The genotype results obtained with the LightCycler protocol were completed within a single working day, whereas those obtained with routine methods required 2–3 days. This method demonstrated 100% reliability in distinguishing wild-type versus mutant alleles. The application of the protocol to screen samples with unknown mutations that have been sequenced allowed the evaluation of the method and highlighted several points. The first point is that all samples with mutations within the gene regions covered by the LightCycler detection probes were positively detected as having a variant allele, which allowed all mutations to be distinguished reliably from wild-type alleles. The second point is that mutations with shifts (e.g. frame shift mutation) or other mutations (e.g. IVS I-5) produced a Tm that was near from the other mutations but distinct from the wild type, thus the identification of a single sample with a B-thalassemia mutation that is uknown indicates that the system can also detect rare or novel mutations so long they lie within the detection sequence of the probes. [20] The identity of results obtained with the newly established method and with our conventional ARMS method in the group of patients, and lack of false positive in the non-thalassemics, clearly confirms the equivalence of the methods for the mutations studied. However, real time method has the advantage of being considerably less time-consuming than ARMS and other conventional methods as sequencing. Thus the major advantage of the proposed method is its simplicity, swiftness and reliability, characteristics that make this method suitable not only for diagnosis but also for epidemiological surveys, which require the analysis of a considerable number of samples. Mutation characterization in carriers is a prerequisite when offering prenatal diagnosis to couples at risk for having an affected child and frequently needs to be achieved as quickly as possible, especially when at-risk couples present after establishment of a pregnancy. [21] References
Copyright 2006 - African Forum for Health Sciences The following images related to this document are available:Photo images[jh06031f3.jpg] [jh06031f1.jpg] [jh06031f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}