 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Medicine and Biomedical Research, Vol. 3, No. 1, June 2004, pp. 7-20 Review Articles An overview of site-specific delivery of orally administered proteins/peptides and modelling considerations Ikhuoria M Arhewoha and Augustine O Okhamafea aDepartment of Pharmaceutics and Pharmaceutical
Technology, Faculty of Pharmacy, University of
Benin, Benin City, Nigeria.
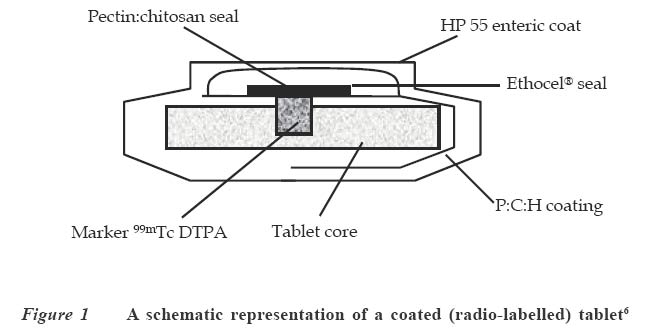
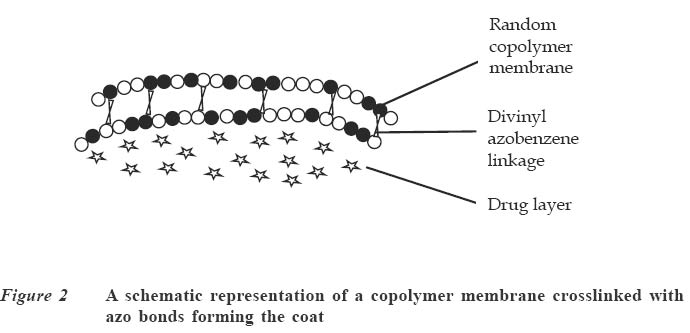
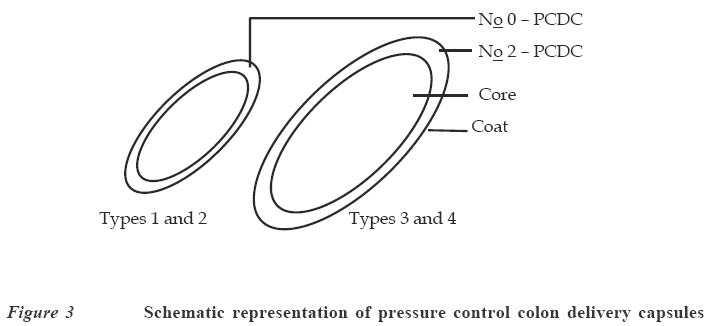
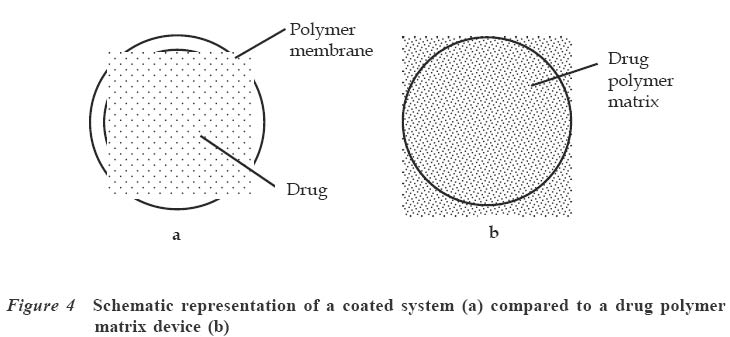
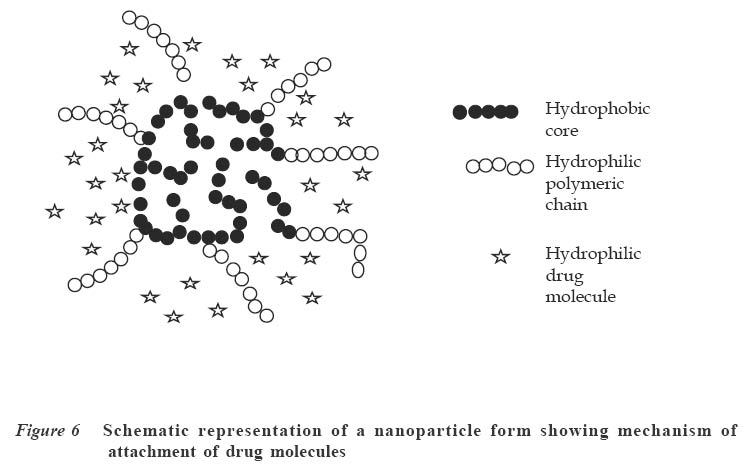
Code Number: jm04002 ABSTRACT Oral delivery of proteins and peptides poses one of the greatest challenges in controlled drug delivery due to degradation by proteolytic enzymes, poor membrane permeability and large molecular size. Therapeutic proteins/peptides are useful in correcting metabolic disorders (e.g., insulin in diabetes mellitus), as antineoplastic agents (e.g., cyclosporin A) and in hormone therapy (e.g., leutinizing hormone releasing hormone analogs in endometriosis and uterine fibroids). Thus, the importance of determining absorption efficiency of peptide and protein drugs is an important consideration. In oral peptide administration, the ideal delivery system is one that releases its contents only at the target region of the gastrointestinal tract (GIT) independent of patient variables while retaining the protein/peptide drug and their absorption promoters at the site of optimal absorption long enough to ultimately achieve maximum therapeutic benefit and safety. Various systems for achieving site-specific delivery of orally administered protein/peptide drugs have been developed in recent years including those based on pH changes, enzymatic activity of intestinal microflora, etc. This article discusses some of these systems including their relative merits and drawbacks. Some relevant mathematical models to resolve release patterns are also considered. Key Words: Site-specific delivery, colon-specific delivery, proteins/peptides, oral administration, modelling INTRODUCTION Drug absorption via the oral route The oral route is often the preferred route for drug administration because it is more convenient for self-administration, allows a wide range of dosage adjustment and drug administration, and can be terminated where untoward effect occurs. However, parenteral administration may be favoured where the drug suffers a very high first pass effect, a quick onset of action is required, the patient is unconscious or cannot swallow, or where the drug may be affected by proteolytic enzymes and acids in the stomach. For drugs of large molecular size such as proteins/peptides and other macromolecules, passage through the walls of the GIT still remains one of the major hurdles to be surmounted before they can produce systemic effect after oral administration. While efforts have been concentrated in the past on absorption of drugs from the stomach, duodenum and the upper part of the small intestine (in the case of enteric coated drugs), only recently has it emerged that specific sites exist in the distal parts of the small intestine and the colon where large drug molecules can be absorbed into systemic circulation if only they can get there.1 Of particular interest in this regard are proteins and peptides. This group of drugs is usually administered parenterally. However, some obvious disadvantages are associated with parenteral administration of drugs. These include poor patient acceptance, invasiveness and cumbersomeness, since several drugs have to be administered repeatedly and over a long period of time. Furthermore, injections are costly and peptides generally display a short half-life;2 hence, frequent administration to maintain adequate blood levels is often required. The need to explore various means possible for achieving oral delivery of these products has, therefore, become imperative. Any realistic attempt to deliver proteins and peptides through the oral route must first take into account the possible losses that could occur as a result of degradation by acids and enzymes in the stomach and small intestine as well as the physicochemical properties of the drugs themselves. The properties include: • Large molecular size • Vulnerability to proteolytic attack • Adherance to surfaces • May be immunogenic and non-biocompatible • Exhibit non-linear pharmacokinenitics • Tend to undergo aggregation and denaturation.2 The release of drug from any dosage form is of great importance as it is a necessary step to determining how much of the drug is available for absorption.3 When formulating a controlled release dosage system for proteins/peptides, for example, it will be imperative to develop a suitable mathematical model for the drug release. This will be useful in predicting the release profile and allow the scientist to adjust release parameters to suit a particular need.3 It has been demonstrated that peptides can be protected effectively from enzyme and acid attack when administered orally by coating with certain polymers.4-6 Furthermore, since it is now known that there are sites in the small intestine and colon where absorption of these drugs can occur,5,6 the possibility of achieving delivery of peptides by the oral route is increasingly becoming a reality. This paper presents an overview of the efforts towards the development of an efficient orally administered protein and peptide drug delivery system and some modelling considerations. THERAPEUTIC PROTEINS AND PEPTIDES Structure Proteins are macromolecules containing fundamental units of any of twenty known α-amino acids in varying numbers and arrangements. Besides the difference in size from conventional molecules, the protein drug has a biological activity that is dependent on its complex structure. The protein molecule has different levels of organisation within each molecule, namely, the primary, secondary, tertiary and quaternary structures, which are responsible for the biological effectiveness and specificity of the actions of proteins. Hence, there is a close relationship between the molecular three-dimensional structure and efficacy of a protein.4 Therefore, it is essential to preserve the structural integrity through all the formulation steps of a delivery system and while the drug is released from the dosage form at the site of delivery. Production Proteins are produced naturally in most living organisms through a process of protein synthesis involving DNA and RNA. Two types of proteins are produced, namely, insoluble or fibrous proteins and soluble proteins. Insoluble proteins are usually isolated from their natural source by removing contaminating materials by means of a suitable array of solvents.7 Appropriate enzymes are used to break down the disulphide bridges/linkages. However, most protein/peptide drugs are soluble proteins, which are obtained first as crude extracts in aqueous solution from their natural source (e.g., insulin from bovine pancreas) from where they are subjected to different filtration and purification stages7 such as dialysis to remove contaminating solute, precipitation with salt or organic solvent. Lyophilization technique is used to obtain the protein, and finally chromatographic procedure (high performance liquid chromatography) is used for purification and to separate the different proteins.7,8 The current trend, however, is the production of proteins by recombinant technology, which has made it easier, faster and produces purer proteins.4,8 Clinical uses A number of protein/peptide drugs already exist and are in the market as parenteral preparations.4 Some of these include leutinizing hormone releasing hormone analogs (leuprorelin acetate and goserelin acetate), which are used in the treatment of endometriosis, uterine fibroids, and prostate and ovarian cancer. Goserelin acetate, which is a decapeptide, is formulated as an implantable device. Bovine somatotropin or bovine growth hormone is used to increase milk production in dairy cattle while recombinant growth hormone is used to treat short stature resulting from growth hormone deficiency in children, and human insulin (a product of recombinant technology) is used to treat type 1 diabetes mellitus.4 DEVELOPMENT AND EVALUATION OF COLON-SPECIFIC DELIVERY SYSTEMS Site-specific delivery to the colon has two major advantages for the development of pharmaceutical products. 1. Treatment of local conditions or colonic diseases such as Crohn's disease, ulcerative colitis, irritable bowel syndrome and colon cancer. 2. Improved absorption of difficult drugs such as peptides and proteins. The ability to deliver such compounds orally can be of great importance. The colon is often identified as a preferred site for drug absorption because it has slow transit, low volume and lacks vigorous stirring, thus creating favourable local conditions for stabilisation and absorption enhancement.9,10 It also lacks the presence of digestive enzyme. The optimal site for delivery in the colon is considered to be the caeccal or ascending colonic regions, thus allowing maximum opportunity for absorption or local action. The different means by which specific delivery can be achieved through the colon include: Systems based on pH change PH-sensitive coatings have been used to achieve site-specific delivery to the small intestine. However, by applying more coatings and/or raising the threshold pH at which dissolution of the coating begins, it is possible to achieve colon-specific delivery by use of enteric polymers.6,10 For instance, tablets containing mesalazine and coated with eudragit S100, which dissolves above pH 7, are marketed in a number of countries. Although this formulation is generally successful in achieving site-specific delivery to the colon, failure of the coating to dissolve has been reported, with patients observing intact tablets in their stools. Mesalazine tablets coated with Eudragit L100, which dissolve at a pH of 6, are also commercially available.11 Scintigraphic assessment indicated that in a group of 13 patients, more than 70% of the tablets disintegrated in the upper large intestine. On the average, disintegration occurred 3.2 hours after gastric emptying. Enteric coating is one of the simplest technologies available for colon specific delivery; it also offers an advantage in terms of cost and ease of manufacture. A major drawback, however, is that the site of drug release from dosage forms coated with a pH-dependent polymer can vary considerably with the pH of the GIT and the thickness of the applied coat.9,11 Macleod et al6 assessed the potential of pectin/chitosan/hydroxyl propylmethyl cellulose phthalate (P: C: H) films for colonic drug delivery. Radio labelled 99mTc-tablets were coated with a P: C: H film in the ratio 3:1:1 (Figure 1) and administered to human volunteers. The gastrointestinal transit of the tablets was assessed by gamma scintigraphy. The results showed that in all cases, the tablets were all able to pass through the stomach and small intestine intact. Break-up of the tablets commenced once they were in the colon, due to degradation of the coat by colonic bacteria. This study highlights the potential of this coating system for colonic drug delivery. Systems based on the enzymatic activity of the intestinal microflora The colonic regions have a high presence of microbial anaerobic organisms providing reducing conditions. Thus, the coating may suitably comprise a material that is redox sensitive.12 Such coatings typically consist of azo-polymers, which can, for example, be composed of a random co-polymer of styrene and hydroxyethyl methacrylate cross-linked with divinyl azo-benzene synthesised by free radical polymerisation (Figure 2). The azo-polymer is broken down enzymatically and specifically in the colon delivery system where peptide drugs are delivered by polymers cross-linked with an azo-aromatic group. When peptide drugs are coated with these polymers, they are protected against gastric and intestinal enzymes. The drugs are subsequently released in the large intestine where enzyme activity is low and the azo bond is cloven by only microbial enzymes in the colon. In vitro evaluation of azo-containing polysaccharide gels, more specifically azo-inulin and azo-dextran gels, for colonic delivery was carried out by Stubbe et al.12 The study evaluated whether in vitro azo-polysaccharide gels can be degraded through both reduction of the azo group in the cross-links as well as enzymatic breakdown of the polysaccharide backbone. The azo-polysaccharide gels were synthesised by radical cross-linking of a mixture of methacrylated inulin or methacrylated dextran and NN-bis-(methacryloylamino) azo-benzene (B(MA)AB), and were characterised by dynamic mechanical analysis and swelling measurements. Azo-dextran gels could be obtained from methacrylated dextran having low degrees of substitution but not from lowly substituted methacrylated inulin. Increasing the amount of B(MA)AB resulted in denser azo-inulin and azo-dextran networks. Compared with their swelling in dimethyl formamide, all azo-dextran gels became more swollen in water while azo-inulin gels shrank upon exposure to water, indicating a more hydrophobic character of the azo-inulin gels. Breakdown of the inulin and dextran chains by inulinase and dextranase, respectively, were observed. However, the degradation of azo-dextran gels by dextranase seemed to be more pronounced than the degradation of the azo-inulin gels by inulinase. The drawback of this system is that long and expensive toxicological studies have to be undertaken before using these new polymers as materials recognised as safe.11 Intestinal pressure-controlled colon delivery capsules These pressure-controlled colon delivery capsules (PCDCs) were prepared by a mini pharmaceutical coating machine (Hi-coater) and the colon delivery efficiencies were evaluated in human subjects by Kimura et al. 13 Caffeine powder, used as a model drug, was suspended in a polyethylene glycol (PEG) 1000 suppository base at 500C and was hardened in number 0 and number 2 size capsular shapes (Figure 3). The capsule-shaped suppositories were coated with 5% ethanolic ethyl cellulose solution using the coating machine. By increasing the coating weight of ethyl cellulose from 28.6 ± 1.1mg to 45.3 ± 0.2mg, the mean coating thickness of number 0-PCDCs increased from 56 ± 1 micron to 64 ± 1 micron. With number 2-PCDCs, the mean coating thickness increased from 50 ± 1 micron to 57 ± 1 micron by increasing the coating weight of ethyl cellulose. Coating membrane thickness of 56 ± 1 micron (type1) and 64 ± 1 micron (type 2) as well as number 2 PCDCs having thickness of 50 + 1 micron (type 3) and 57 ± 1micron (type 4), were evaluated in vivo in man. Following oral administration of test preparations containing 75mg of caffeine, saliva samples were obtained and salivary caffeine levels were measured by an HPLC method. The first appearance time, T1, of caffeine in the saliva was used as a parameter for the estimation of the release times of caffeine from the PCDCs in the gastrointestinal tract. The results obtained for the T1 values are shown in Table 1. There were good correlations between ethyl cellulose coating membrane thickness and in vivo T1 values. A colon arrival time of five hours was reported in the subjects by gastrointestinal magnetomarkergraphy. PCDCs having a mean coating thickness of 64 ± 1 micron for number 0 capsules and of 57 ± 1 micron for number 2 capsules were thought to deliver to the human colon efficiently. Matrix tablets Unlike the coated systems, where drug release is determined by the thickness of the coat and dissolution of the polymer membrane, drug release in the matrix system is determined by the dissolution rate of the polymer. Some workers developed colon-targeted drug delivery systems for mebendazole using guar gun as a carrier.14 Matrix tablets containing various proportions of guar gum were prepared and the tablets evaluated for drug content uniformity and subjected to in vitro drug release studies. The amount of mebendazole released from the matrix tablets at different time intervals was estimated. Guar gum matrix tablets released 8-15% of the mebendazole in the physiological environment of the stomach and small intestine depending on the proportion of guar gum used in the formulation. When the dissolution study was continued in simulated colonic fluids, the matrix tablets containing 20% guar gum released another 83% of mebendazole after degradation. The matrix tablets containing 30% guar gum also released about 50% of mebendazole in simulated colonic fluids, indicating the susceptibility of the guar gum formulations to the rat caecal contents. The results of the study show that the matrix tablets containing either 20% or 30% of guar gum are most likely to provide targeting of mebendazole for local action in the colon. (Figure 4) Conjugate or complex formation (pro-drug approach) The release of drugs can also be modified to control absorption at colonic sites. Yano et al15 designed a colon-specific delivery of prednisolone appended to cyclodextrin to form a conjugate. Prednisolone, which is a typical glucocorticoid, has been widely used for the treatment of inflammatory bowel disease (IBD). However, when prednisolone is administered orally, a large amount of the drug is absorbed from the upper GIT and causes systemic side effects. In this study, the anti-inflammatory and systemic side effects of the prednisolone succinate cyclodextrin ester conjugate after oral administration were studied in rats with IBD. The anti-inflammatory effect of the conjugate was comparable to that of prednisolone alone. On the other hand, the low systemic side effects of the conjugate were attributable to passage of the conjugate through the stomach and small intestine without the drug being absorbed in significant amounts. The results suggested that predisolone conjugate is useful as a delayed-released type pro-drug of prednisolone for colon specific delivery owing to alleviation of the systemic side effects while maintaining the therapeutic effect. Figure 5 is an example of a pro-drug of doxorubicin conjugated with a polyethylene glycol 1000 (PEG 1000) and poly (α,β-aspartic acid) block copolymer.16 The general strategy of pro-drug technology, which has been known and practiced for decades, is to synthesise the chemically modified version of the parent drug that overcomes the inherent problem in the ability of the drug to be adequately absorbed. For instance, a drug may be too hydrophilic to partition into biological membranes. If a pro-drug can be synthesised that modifies the parent drug partitioning behaviour appropriately, then flux across a biological membrane can be achieved. Inherent in this whole approach is the feature that the pro-drug must be readily reversible to the parent drug either by hydrolytic or enzymatic mechanism, and that the regenerated parent drug must retain its intrinsic pharmacological activity. Kopeck17 developed a system for attachment in the colon, i.e., muco-adhesive oral drug delivery system (MADS). The system is based on N-(2-hydroxy propyl) methacrylamide co-polymer with approximately 3mol% of side chain terminated in fucosylamine. The colonic association of this copolymer was compared with its counterpart devoid of fucosylamine residues in guinea pigs. The colonic association was found to be time-dependent. Twenty four hours after oral administration of radiolabeled co-polymers, the polymer containing fucosylamine showed twice as much colonic adhesion as that devoid of these moieties. At the end of 48 hours, although the levels of colon-association radioactivity were decreased for both polymers, the co-polymer with fucosylamine moieties exhibited significantly higher association with the colon. In order to further improve colon specificity of this bio-adhesive delivery system, an azo linkage specifically reduced by colonic bacteria was employed to attach 5-aminosalicylic acid, a drug useful in the treatment of ulcerative colitis, to the polymer backbone. Incubation of this delivery system with Streptococcus faecium, a strain of bacteria commonly found in the colon, resulted in the reduction of the azo bond, followed by the release of 5-aminosalicylic acid. Based on these observations, it was suggested that site-specific delivery to the colon could be achieved by conjugating a drug through a linkage specifically cleaved in the colon to a bio-adhesive polymer with adhesive moieties specific for the colon.18 Nanoparticles Nanoparticles are expected to become drug carriers for achieving oral peptide delivery. Different materials have been used to prepare the nanoparticulate systems. Convereur and Puisier 19 used polyisobutylcyanoacrylate as drug carrier. Poly (N-isopropylacrylamide) hydrogel nanoparticles have also been prepared.20 Shinji et al 21 designed nanoparticles using graft copolymers, which were obtained by copolymerizing macromonomers and other monomers. Poly (N-isopropylacrylamide) and poly (N-vinylisobutyramide) were some of the polymers used as pH sensitive (anionic, cationic and thermosensitive nonionic) hydrophilic polymer chains incorporating the peptide drug while polystyrene was used as the hydrophobic core (Figure 6). The nanoparticles were produced by dispersion copolymerization and their usefulness as carriers that incorporate hydrophilic compounds such as peptide drugs was evaluated. The effect on peptide absorption via the GIT, using Salmon calcitonin (sCT) as a model drug was also studied. The result showed that the rate of sCT incorporation into the nanoparticles was relatively high and depended on the chemical structure of the hydrophilic polymer chains located on the nanoparticle surface. These nanoparticles enhanced the oral absorption of sCT in rats. It was also demonstrated that the absorption enhancement of sCT by these nanoparticles resulted mainly from both mucoadhesion of nanoparticles incorporating sCT in the GIT and increase in the stability of sCT against digestive enzymes. The nanoparticles composed of novel graft copolymers were demonstrated to be useful carriers for oral peptide delivery. MODELLING CONSIDERATIONS Drug delivery devices are devices that are capable of adjusting drug output to meet physiological need. The major problem with the delivery of therapeutic proteins and peptides is that of imparting good bioavailability to the peptide-derived drug while maintaining pharmacological efficacy. A mathematical model is usually applied to describe the release kinetics, understand the release mechanism and to be able to adjust model parameters to achieve particular kinetics.22 Several models have been developed for different systems; in the case of micro-encapsulated systems, there is no single release model that can be conveniently applied to the various types of microcapsules. The release model in most cases will be classified according to their controlling physical mechanisms.23 Diffusion models Diffusion of drugs from polymer-based formulation is usually described conveniently by Fick's first and second laws of diffusion written as follows:
Where C and J are the concentration and maximum flux of the drug, respec-tively; x and t are position and time of release respectively D is the drug diffusion coefficient through the polymer. The negative sign in equation (1) signifies that diffusion occurs in a direction opposite to that of increasing concentration. From equations (1) and (2) above, the following information can be derived: (i) The concentration profile (ii) The rate of release of drug:
(iii) The total amount of drug released per sectional area, which is given by: It is assumed here that the drug exhibits a constant release rate irrespective of the initial loading, that is, diffusion coefficient is not dependent on the concentration of drug. This is zero order release kinetics. Higuchi24 had developed a simple pseudo-steady state model based on Fickian diffusion with zero surface concentration, which gives the amount of drug released and its rate of release from a planar surface according to the following equations:
In the above equations: Mt is the total amount of drug released per unit area at time t D is the diffusion coefficient of the drug in the release medium divided by the tortuosity factor (ô) is the final volumetric porosity of the matrix Cs is the solubility of the drug in the release medium Co is the drug concentration in the matrix t is the time. This should yield a linear concentration gradient in the dissolved region. By solving Fick's second law of diffusion and introducing a moving boundary condition; Paul and McSpadden25 developed the exact analytical solution of the boundary problem for non-porous systems. The amount released is given by: Where
x* is the moving front position Cp is the drug solubility in the polymer D is the diffusion coefficient of the drug in the polymer Miller and Peppas26 gave the corres-ponding solution for porous systems: The moving front position can be determined by solving the following equation:
Where In equation (8), a porosity term with a 2/3 exponent is proposed for the cross-sectional porous area available for diffusion per unit area exposed to the release medium. This two dimensional cross-sectional porosity is identical to the volumetric porosity (e). Therefore, equation (8) was modified to introduce an exponent of 1 to the porosity term thus: Movement of proteins across polymer membrane is by diffusion.27 One possible disadvantage of using microcapsules, however, is the increased transport resistance particularly since the molecules in question (proteins and peptides) are large. The development of mathematical models for protein diffusion across semi-permeable membrane was carried out by Kwok et al.27 The hollow sphere model reported by Carslaw and Jaeger28 was used and Fickian diffusion was assumed. In the study, alginate-poly-L-lysine (PLL) membrane was used and this was justified by the fact that the alginate-PLL membrane used is highly porous with approximately 90% by weight of water (a hydrogel). Hence, with these large pore sizes, the semi-permeability brought about by the ionic groups in the membrane will be negligible when compared to the ordinary Fickian diffusion process. The diffusion of proteins through the membrane was given by: Where r = radial position (direction of diffusion) t = time At the membrane-intracapsular solution interface, negligible resistance was assumed (Case 1). In this consideration, the protein diffuses from the bulk solution into the microcapsule core. In order to successfully develop a model, it is first of all necessary to investigate the possible mechanism of drug release from the microcapsules in vivo. For example, at the pH of 6.5 and above, the chitosan-alginate microcapsule is believed to swell (a hydrogel).29 This will allow the influx of water into the capsule to fill the pores and dilute the drug, thus creating a concentration gradient. The drug then begins to diffuse out at a rate that will be determined by the membrane permeability, drug molecular size and electrostatic repulsion.29 Diffusion of proteins increases with increase in pH because of increased negative charge on the protein. The temperature dependence of the diffusion coefficient follows Arrhenius law. The concentration of chitosan solution is also important, as this will determine the porousity of the capsule shell. In order to analyse drug release data from swelling-controlled release systems, the data is fitted into Equation (12), which is derived from Fick's second law: The value of the exponent n is characteristic of both Fickian or non-Fickian diffusion behaviour of swelling-controlled release systems. It is also possible to derive sufficient and necessary conditions for obtaining zero order release from swelling controlled release systems30 by examining the dimensionless number, Sw, that is, the so-called swelling interface number defined according to the equation below: When Sw <<1, zero order release should be expected, whereas for values of Sw >1, Fickian diffusion is observed. CONCLUSION This paper has reviewed some of the efforts so far made to achieve oral delivery of large drug molecules such as protein/peptide drugs. Therapeutic proteins and peptides are presently used as parenteral preparations, amongst others, in correcting metabolic disorders (insulin, glucagons, calcitonin) as antineoplastic agents (cyclosporin) and in hormone therapy (bovine somatotropin and human growth hormone). The benefits of achieving oral delivery of these drugs are also outlined. Some diffusion-based drug release models have been presented. It is certain that significant progress has been achieved. Research in this direction is continuing at a high pace. It is expected that even the recently emerging therapeutic genes will also be candidates for the oral delivery route. Refrences
© CMS UNIBEN JMBR The following images related to this document are available:Photo images[jm04002t1.jpg] [jm04002f7.jpg] [jm04002f6.jpg] [jm04002f4.jpg] [jm04002f5.jpg] [jm04002f2.jpg] [jm04002f8.jpg] [jm04002f3.jpg] [jm04002f1.jpg] |
| |||||||||
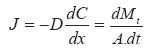 ………………………(3)
………………………(3)
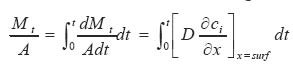 ……. (4)
……. (4)
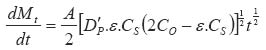 …………(6)
…………(6)
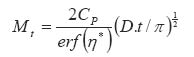 …………………..(7)
…………………..(7)
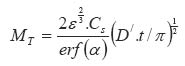 ……………………..(8)
……………………..(8)
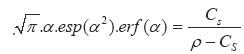 . ...……………. (9)
. ...……………. (9)
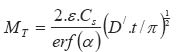 …………………(10)
…………………(10)
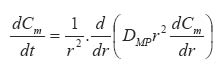 …………….(11)
…………….(11)
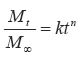 …………………………………..(12)
…………………………………..(12)
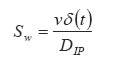 ……………………..(13)
……………………..(13)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}