 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Medicine and Biomedical Research, Vol. 4, No. 1, June 2005, pp. 9-21 REVIEW ARTICLES Current trends in the production and biomedical applications of liposomes: a review MU Uhumwangho and RS Okor Department of Pharmaceutics and Pharmaceutical Technology,
University of Benin, Benin City, Nigeria.
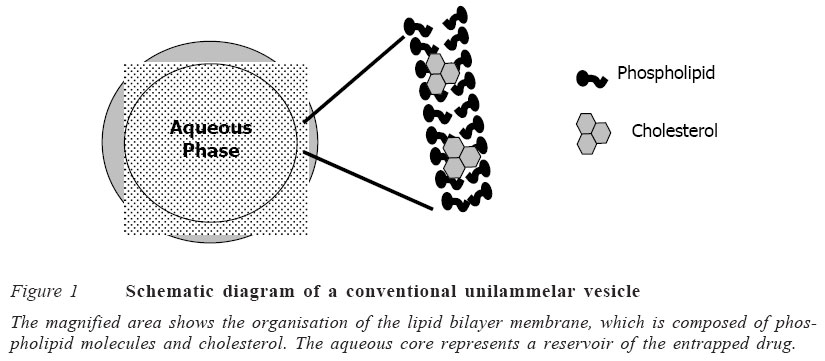
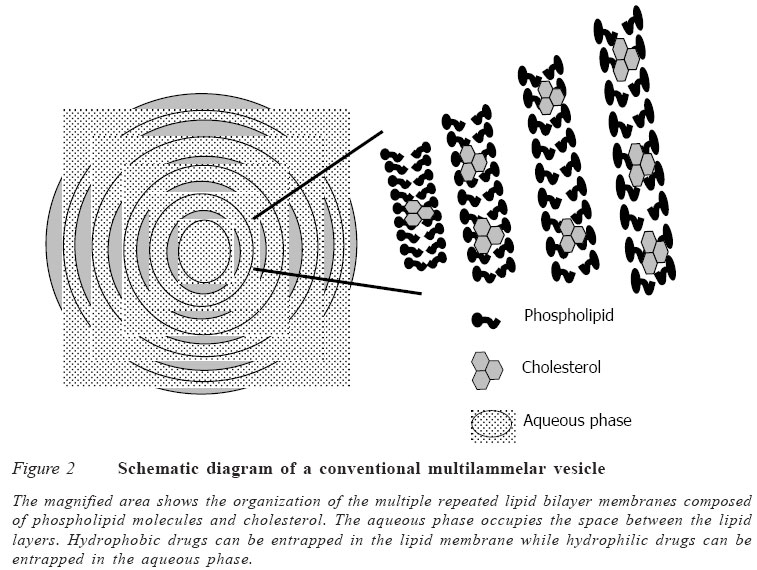
Code Number: jm05002 ABSTRACT A review of literature was carried out to determine methods of production of liposomes, their stability, biodistribution and their uses as drug delivery systems. The conventional method of preparing liposomes is basically for the multilamellar vesicles (MLVs). However, other methods are used to reduce the size of these MLVs to small unilamellar vesicles (SUVs) so as to increase their plasma lifetime and consequently increase the possibility of achieving greater tissue localisation. Some of these methods of size reduction are sonication and high pressure extrusion. Each of these methods has its own advantages and disadvantages. Large unilamellar vesicles (LUVs), on the other hand, are prepared mainly by detergent removal method and reverse phase extrusion technique. There are also improved pharmacokinetic properties with liposomal drugs compared to free drugs, though some formulation factors affect the release kinetics of the liposomal drugs. The review also shows that liposomes have a lot of biomedical applications and uses. They have been used in drug targeting, oral delivery of vaccines, insulins, peptides and some compounds, which are usually degraded in the gastrointestinal tract. It has also found application in topical therapy especially in the eye and lungs. Other areas of application are in cancer chemotherapy and treatment of human immunovirus (HIV) infection. The control of the stability of liposomes is an essential pre-requisite for effective use as drug carriers. Leakage of the liposome is attributable mainly to differences in lamellar structure. For instance, MLVs are less prone to leakage than ULVs. The use of a combination of saturated phospholipid and cholesterol in the formulation of the liposomes has also been found to enhance stability with lower tendency to leakage. KEY WORDS: Liposomes,phospholipid, encapsulation, applications INTRODUCTION Liposome was discovered about 40 years ago by Bangham and co-workers and was defined as microscopic spherical vesicles that form when phospholipids are hydrated or exposed to an aqueous environment.1 Liposomes are microscopic vesicles composed of a bilayer of phospholipids or any similar amphipathic lipids. They can encapsulate and effectively deliver both hydrophilic and lipophilic substances,2,3 and may be used as a non-toxic vehicle for insoluble drugs.4 Weiner et al5 defined liposome as a microstructure consisting of one or more concentric spheres of lipid bilayer separated by water or aqueous buffer compartments (Figures 1 and 2). The typical characteristic of bilayer-forming lipids is their amphiphilic nature: a polar head group covalently attached to one or two hydrophobic hydrocarbon tails. When these lipids, e.g., phosphatidylcholine, phosphatidyl ethanolamine or phosphatidyl glycerol, are exposed to an aqueous environ-ment, interactions between themselves (hydrophilic interactions between polar head groups and van der Waals interactions between hydrocarbon chains and hydrogen bonding with water molecules) lead to spontaneous formation of closed bilayers.6 Liposomes can differ in size, ranging from the smallest vesicle (diameter 20nm) to liposomes that are visible under the light microscope, with a diameter of 1μm or greater, equal to the dimensions of living cells.6 Liposome can carry drugs in one or three potential compartments (water soluble agents in the central aqueous core, lipid soluble agents in the membrane, peptide and small proteins at the lipid aqueous interface). They are classified structurally into multilamellar vesicles (MLVs) and unilamellar vesicles (ULVs).7 ULVs have a single phospholipid bilayer membrane and a diameter of 0.05-0.25μ m. These liposomes (i.e., ULVs) can be further classified into large unilamellar vesicles (LUVs) with a diameter of 0.05-0.25μm and small unilamellar vesicles (SUVs) with a diameter of 0.05-0.10μm. Liposomes are designed in such a way that the solute can be entrapped in the aqueous compartment (polar solute) or embedded in the lipid bilayers (lipohilic or amphiphilic solute). Since ULVs contain a large central aqueous compartment, they are ideally suited for the encapsulation of water soluble agents.8 MLVs are composed of concentric phospho-lipid bilayer membranes in an onion skin arrangement and have a diameter of 1-5μm. ULVs contain a small aqueous compartment (<10%), which means that they preferentially entrap lipid soluble drugs.8 This observation is important to the design of liposomes for cancer therapy since a number of active cytotoxic drugs are highly lipid soluble.9 Liposomes have been studied for many years as carrier systems for drugs,10 with advantages such as enhancement of thera-peutic efficacy at low dosage and, hence, reduction in toxicity of the encapsulated agent; improved pharmacokinetic profiles, e.g., enhanced tissue penetration and increased biological half life; targeting to tumour tissues, e.g., liposomal doxorubicin; and increased stability of the drug particularly against enzymatic degradation.2,11,12 The purpose of this study is to review current research efforts in the technology of liposome production and to review current biomedical applications of liposomes, with a view to stimulating further research in this area of study. Literature search was the major approach for the study.
FORMULATION OF LIPOSOME Liposomes are made from pure lipids or a combination of lipids. The lipids commonly employed in liposome formulations are phospholipids. Liposomes have been prepared from a variety of synthetic and naturally occurring phospholipids, they generally contain cholesterol.13 The incorporation of cholesterol into the lipid bilayer membrane generally enhances the stability of liposomes in serum, reduces the permeability of the membranes to water soluble molecules and increases the fluidity or microviscosity of the bilayer.5,14 The most commonly used phosholipids in liposome preparation are: egg phosphatidylcholine, synthetic dipalmitoyl-DL-α-phosphatidylcholine, brain and synthetic phosphatidylserine, sphingomyelin, phosphatidylinositol and ovolecithin. Usually, a zwitterionic or non-ionic lipid is used as the basic lipid for the preparation of liposomes. The net surface charge of liposome can be modified by the incorporation of positively charged lipids such as stearylamine, or negatively charged lipids such as diacetylphosphate, phosphatidyl glycerol or phosphatidyl serine.6 The presence of negatively or positively charged lipids lead to a greater overall volume for aqueous entrapment and reduces the likelihood of aggregation after preparation of the liposomes.15 Cationic liposomes display some disadvantages such as cytolytic and cytotoxic activities. Yoshihara and Nakae16 have demonstrated that cationic liposomes containing stearylamine showed an in vivo toxicity in rabbits. This effect was attributed to haemolysis of the erythrocytes and was directly related to the amount of stearylamine present in the liposome composition.
Formulation factors affecting the degree of drug entrapment The extents of drug entrapment and retention as well as factors influencing them are important considerations in the design of liposome-mediated drug delivery systems. Drugs may be entrapped in the aqueous and/or lipid phase of the liposome.
a. Aqueous entrapment This relates to the aqueous volume in the liposome. The larger the aqueous volume the greater the amounts of polar drugs that can be encapsulated.17 Multiple compartment liposomes encapsulate higher percentages of aqueous soluble drugs than single compart-ment vesicles, because of the larger volume of encapsulated aqueous space in the former. Formulations that promote formation of MLVs are thus associated with higher aqueous entrapment. Osmotic swelling and/or incor-poration of charged lipids, e.g., phosphatidy-lserine into bilayers are measures for increasing the aqueous volume in liposomes.5,15 The latter is due to charge repulsion separating adjacent bilayers, resulting in increases in trapped aqueous volume. Aqueous solubility of the drug is another factor, hence, the extent of drug entrapment in liposomes (MLVs) can vary markedly as seen in the following examples: 2.2-8.4% for penicillin, 2.3-11.6% for actinomycin D, 18% for methothrexate and up to 60% for bleomycin. Leakage of entrapped solute is another formulation problem. Cholesterol modifies the fluidity of lipid membranes, thereby influencing the degree of retention of drugs by vesicles as well as stabilising the system against enzymatic degradation.5 Large molecules (e.g., peptides and proteins) are better retained than smaller molecules, which can diffuse slowly through the lipid layers.
b. Lipid entrapment Lipid soluble drugs are entrapped in the lipid layers of liposome. Here, the entrapment efficiency can be as high as 100%, irrespective of liposomal type and composition. An example of a drug that is hydrophobic in nature is camptothecin.18 The retention of such hydrophobic drugs is also high when the liposomes are placed in aqueous biological environment because of their high lipid-water partition coefficients.
Formulation factors affecting stability of liposomes The stability of liposomes refers to their ability to retain entrapped solutes, chemical stability of both the entrapped solutes and the lipid membranes. Solute leakage depends on membrane permeability and on the interaction with components of biological fluids. Membrane fluidity can be controlled to reduce leakage by supplementing the lipid bilayer with cholesterol14 or by manipulating the hydrophobic/lipophobic character of the bilayers, for example, with the use of fluorinated lipid.19,20 The rate of solute leakage also depends on the lamellar structure of liposomes, for instance, MLVs are less prone to leakage than ULVs.21 In order to minimise leakages, liposomes are stored in the form of freeze-dried powders. The lipid vesicles can undergo chemical degradation. For instance, phospholipids can be hydrolysed to lysophospholipids, which are also subject to further hydrolysis.22 The 2- lysophospholipids are the main initial products of hydrolysis.23 Hydrolytic degra-dation of either the lipid or entrapped drug may be pH related but can be prevented or minimised by freeze-drying of liposome suspension to dry powders. When unsatu-rated phospho-lipids are used to prepare liposomes, special precautions must be taken to minimise oxidation. These include the use of light resistant containers, use of antioxi-dants such as α-tocophenol, de-aeration with argon or nitrogen to minimise exposure to oxygen, and removal of heavy metals from the preparation.
Technology of liposomes production Since the early 1970s many hundreds of drugs, including anti-tumour and antimi-crobial agents, chelating agents, peptide hormones, enzymes, other proteins, vaccines and genetic materials, have been incorporated into the aqueous or lipid phases of liposomes of various sizes, compositions and other characteristics by an ever-increasing number of techniques. Liposomes have evolved from mere experimental tools of research to industrially manufactured products for clinical and veterinary use. This success depends on advanced techniques to obtain efficient drug entrapment and increased stability of the products. The conventional method and the advanced techniques based on this method are discussed as follows.
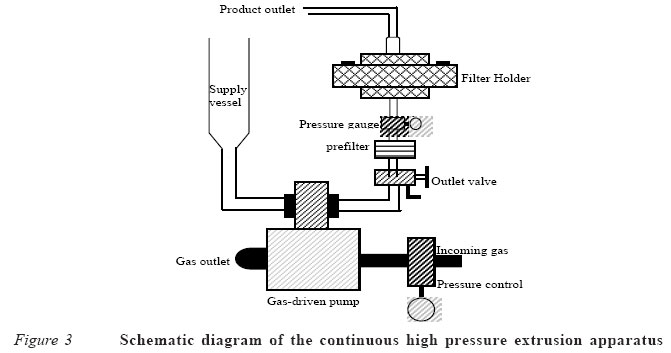
Convectional method The convectional method was first described in detail by Bangham et al1 for the preparation of MLVs. In the procedure; the phospholipids are dissolved in an organic solvent (usually a chloroform/methanol mixture) and deposited from the solvents as a thin film on the wall of a round bottom flask by use of rotary evaporation under reduced pressure. MLVs form spontaneously when an excess volume of aqueous buffer containing the drug is added to the dried lipid film. Drug containing liposomes can be separated from non-sequestered drug by centrifugation of the liposomes or by gel filtration. The time allowed for hydration of the dried film and conditions of agitation are critical in determining the amount of the aqueous buffer (or drug solution) that will be entrapped within the internal compartments of the MLVs. For instance, it is reported that more of the aqueous phase can be sequestered when the lipid is hydrated for 20 hours with gentle shaking, compared with a hydration period of two hours, with vigorous shaking of the flask, even though size distribution of the MLVs was unaffected.24 This means that slow hydration is associated with greater entrapment of aqueous volume. Sonication method This method is used in the preparation of SUVs25 and it involves the subsequent sonication of MLVs prepared by the convectional method1 either with a bath type or a probe type sonicator under an inert atmosphere, usually nitrogen or argon. The principle of sonication involves the use of pulsed, high frequency sound waves (sonic energy) to agitate a suspension of the MLVs. Such disruption of the MLVs produces SUVs with diameter in the range of 15-50nm. The purpose of sonication, therefore, is to produce a homogenous dispersion of small vesicles with a potential for greater tissue penetration. The commonly used sonicators are of the bath and probe tip type. A probe tip sonicator delivers high sonic energy to the lipid suspension but has the disadvantage of overheating the lipid suspension to cause degradation. The probe tip also tends to release titanium particles into lipid suspension, which must be removed by centrifugation. For this reason, bath sonications are the more widely used. By this technique, a test tube containing the suspension is placed in the bath sonicator and sonicating for 5-10 minutes above the transitional temperature of the lipid (i.e., the temperature at which the lipid melts). Comparatively, sonication with a probe results in faster breakdown of the MLVs to smaller structures than can be achieved by a bath sonication. The reduction in size of the liposomes, however, also decreases the amount of interior aqueous space, thereby limiting the amount of water-soluble drugs that can be entrapped. However, degradation of lipids, metal particle shedding from the probe tip (titanium particles) and generation of aerosols from solutions containing radioactive traces, carcinogenic chemicals or infectious agents that have been added to the preparation by probe technique could cause serious biohazards, which are less frequently encountered with bath sonication. Bath sonication is a closed system allowing for temperature control to minimise thermo-degradation of the lipid and entrapped substance. The position of the tube and water level in the bath is also regulated for maximum efficiency. The major drawbacks in the preparation of liposomes by sonication include oxidation of unsaturated bonds in the fatty acid chains of phospholipids and hydrolysis to lyso-phospholipids and free fatty acids. Another drawback is the denaturation or inactivation of some thermolabile substances (e.g., DNA, certain proteins, etc) to be entrapped. High-pressure extrusion method This is another method for converting MLV to SUV suspensions. By this method, suspensions of MLVs prepared by the convectional method1 are repeatedly passed through filters polycarbonate membranes with very small pore diameter (0.8-1.0μm) under high pressure up to 250psi.24 By choosing filters with appropriate pore sizes, liposomes of desirable diameters can be produced.24,26,27 The mechanism of action of the high pressure extrusion method appears to be much like peeling an onion. As the MLVs are forced through the small pores, successive layers are peeled off until only one remains. Besides reducing the liposome size, the extrusion method produces liposomes of homogeneous size distributions.24,28 A variety of different lipids can be used to form stable liposomes by this method.27,29 Extrusion at low pressures <1Mpa is possible when lipid concentration is low,30 but the most commonly used pressures are about 5Mpa.28,30 The method is amenable to small scale production only. In order to overcome this problem, Schneider and co-workers31 designed and tested a new continuous extrusion apparatus, which operates at high pressures of up to 10.5Mpa with the advantage of a high output. The schematic diagram of the continuous high-pressure extrusion apparatus is shown in Figure 3. Solubilisation and detergent removal method This method is used in the preparation of LUVs32 and it involves the use of detergent (surfactant) for the solubilisation of the lipids. Detergents used include the non-ionic surfactants [e.g., n-octyl-bete-D-glucopyranose (octyl gluside), anionic surfactants (e.g., dodecyl sulphate) and cationic surfactants (e.g., hexadecyltrimethyl ammoniumbromide). The procedure involves the solubilisation of the lipids in an aqueous solution of the detergent and the protein(s) to be encapsu-lated. The detergent should have a high critical micelle concentration (CMC), so that it is easily removed. The detergent is subsequently removed by dialysis or column chromatography.5 During detergent removal, LUVs of diameter 0.08-0.2μm are produced. This detergent removal method has been found suitable for the encapsulation of proteins of biomedical importance.6 Reverse phase evaporation technique Szoka and Papahadjopoulos33 pioneered the preparation of lipid vesicles by reversed phase evaporation technique. It consists of a rapid injection of aqueous solution of the drug into an organic solvent, which contains the lipid dissolved with simultaneous bath sonication of the mixture leading to the formation of water droplets in the organic solvent (i.e., a "water-in-oil" emulsion). The resulting emulsion is dried down to a semi solid gel in a rotary evaporator. The next step is to subject the gel to vigorous mechanical agitation to induce a phase reversal from water-in-oil to oil-in-water dispersion (i.e., an aqueous suspension of the vesicles). During the agitation, some of the water droplets collapse to form the external phase while the remaining portion forms the entrapped aqueous volume. Large unilamellar vesicles (diameter 0.1-1μm) are formed in the process. This method has been used to encapsulate both small and macromolecules such as RNA and various enzymes without loss of activity. The expected limitation of this method is the exposure of the material to be encapsu-lated to organic solvents and mechanical agitation, which can lead to the denaturation of some proteins or breakage of DNA strands. Reports of such limitations are however rare in the literatures. Storage of liposomes: freeze-drying Liposome dispersions are potentially prone to hydrolytic degradation and leakage. Hence, it is desirable to freeze-dry the suspension to a powder and store in this dried form.34 The powder can be reconstituted to an aqueous suspension immediately before use. By doing so, SUVs may be converted to MLVs dispersion upon rehydration. Addition of a carbohydrate (trehalose) during freeze-drying prevents fusion and leakage of the vesicles.35 Pharmacokinetic considerations Most small molecular chemotherapeutic agents have a large volume of distribution on intravenous (IV) administration of liposomes.36 The result of this wide distribution is often a narrow therapeutic index due to a high level of toxicity on healthy tissues. Through encapsulation of drugs in liposomes, the volume of distribution is significantly reduced and the concentration of drug at the desired site of action increased. For instance, liposomal drug delivery led to an increase in the amount of drug that can be effectively delivered to tumour sites in anticancer therapy.37,38 Liposomes are predominantly removed from circulation by phagocyte cells of the reticuloendothelial system (RES), thus accumulating to a large extent in organs like liver and spleen. This biodistribution pattern can be used for passive targeting of diagnostics to these organs. The RES should, therefore, be saturated with empty vesicles when other sites are the drug targets. Information on biodistribution is, therefore, important for drug targeting by liposomes. Liposomes given intravenously usually interact with at least two distinct groups of plasma proteins.39 These are the plasma high density lipoproteins and the so-called opsonins, which bind to the surface of vesicles and mediate their endocytosis by the mononuclear phagocyte system (macrophages). The rate of liposome clearance from blood circulation will, therefore, depend on the ability of opsonins to bind to the liposome surface. The rate can be manipulated through appropriate selection of liposome charac-teristics.40 For instance, "fluid" vesicles are removed more rapidly from blood circulation than "rigid" ones. Clearance from the blood stream is also influenced by vesicle size and surface charges.41 The longest half-life is obtained when liposomes are relatively small (diameter <0.05μm) and carry no net surface charge.6 The pharmacokinetic behaviour of liposomes depends on the route of injection42 such as intraperitoneal, subcutaneous or intramuscular route. Lasic and Papahad-jopoulos43 have shown that coating the liposome surface with polyethyleneglycol and other hydrophobic part of phospholipids44 substantially prolongs the half-life of liposomes in the blood.
Biomedical applications of liposomes Both hydrophilic and hydrophobic drugs can be encapsulated in liposomes.2-4,45 Liposomes are also relatively non-toxic and biodegra-dable.46 They therefore have a wide range of biomedical applications. Protection against enzymatic degradation of drugs Liposomes are used to protect the entrapped drug against enzymatic degradation whilst in circulation.1 The basis is that the lipids used in their formulation are not susceptible to enzymatic degradation; the entrapped drug is thus protected while the lipid vesicles are in circulation in the extracellular fluid. Upon penetration into the cell, the entrapped drug is released either by diffusion through the microsphere shell, dissolution of the shell or degradation of the shell by lysosomal enzymes. Thus, β-lactamase sensitive antibiotics, e.g., the penicillins and cephalosporins have been encapsulated due to this reason to protect against the β-lactamase enzyme. Liposomes also offer protection for its encapsulated drugs in the environment of the gastrointestinal tract47 and facilitate the gastrointestinal transport of a variety of compounds.48,49 Liposomes are therefore candidates to be explored for oral delivery of insulin and proteins for use as vaccines, which are otherwise orally degradable. Liposomes offer a number of advantages as carriers of vaccine agents40 in that they are biodegradable and non-toxic. Drugs encapsulated in lipososmes can elicit both humoral immunity when given orally and cell-mediated immunity.50 Liposomes are now being employed as oral vaccines in numerous immunization procedures.6 Twenty five years after the discovery of the immunological adjuvant properties of liposomes, they are now considered the major candidate as the base for oral vaccine against hepatitis A, which is being licensed for use in humans.51
Drug targeting The need for "drug targeting" arises from a problem situation whereby a drug adminis-tered (iv for example) enters the blood stream and is distributed to varying extents throughout the body when the actual desire is to deliver or direct the drug selectively to its site of action. This site could be an organ structure, a cell subset, or even an intra-cellular region. In such a case pumping the drug throughout the whole body is not only wasteful but, more fundamentally, it is also likely to lead to undesirable side effects. On the other hand, restricting the distribution of the drug to the specific target site should allow for an increase in efficacy at low dose with attendant decrease in toxicity.2,46,52 Hence, the benefits of drug targeting include reduced drug waste, and it is possible to deliver a drug to a tissue or cell region not normally accessible to the free or untargeted drug. The approach for drug targeting via liposomes involves the use of ligands (e.g., antibodies, sugar residues, apoproteins or hormones), which are tagged on the lipid vesicles. The ligand recognises specific receptor sites and, thus, causes the lipid vesicles to concentrate at such target sites. By this approach the otherwise preferential distribution of liposomes into the reticuloen-deothelial system RES (liver, spleen and bone marrow) is averted or minimised. The preferential distribution of liposomes into the RES can be modified by the incorporation in the liposome membrane of protein or carbohydrates possessing specific affinity toward a target tissue or organ.45,53,54 A ligand selection is based on its recognition by, and specificity for, the target site. In cancer treatment, for example, one of the approaches is to target the drug to tumour cells via receptor specific ligands, which may be specific antibodies for antigens produced by the tumour cells. The first step, therefore, is to determine the antigens that are produced by the tumour cells. Also, molecules bearing oligosaccharide chains have been used as ligands for direction, and specific attachment, to ganglion sites in cells.55 Topical drug delivery The application of liposomes on the skin surface has been proven to be effective in drug delivery into the skin.56-59 Liposomes increase the permeability of skin for various entrapped drugs and at the same time diminish the side effect of these drugs because lower doses are now required.60,61 Liposomes have also found an important application in cosmetics for skin care preparations.62 In this regard, the liposomes are applied to the skin in the form of solution or in hydrogels.63 Hydrophilic polymers are suitable thickening agents for the gels. However, the liposomes may in certain instances be trapped in the polymeric network of the hydrogels and, hence, impair bioavailability into the skin.64,65 Nevertheless, Gabrijelcic et al66 found enhanced transport of liposome-entrapped substances into the skin from hydrogels prepared from xanthan gum. The enhanced drug transport into the skin is attributed to the lipid nature of the vesicles, which serve as carriers for the drug.
Treatment of human immunodeficiency virus (HIV) infections Several antiretroviral nucleotide analogues have been developed for the treatment of patients suffering from the acquired immunodeficiency syndromes (AIDS). These include antisense oligonucleotide, which is a new antiviral agent that has shown potential therapeutic application against HIV-1.67 These antiviral agents are able to combat replication of the HIV by inhibiting reverse transcriptase and, thereby, viral DNA synthesis.68 However, these agents display a dose-related toxicity. The effective dose can be reduced by encapsulation of such drugs in liposomes, thus reducing the incidence of toxicity. The greater efficacy of the liposomal formulation relates to the preferential uptake of the liposomes into the virus compared with the host tissue.69 Enhanced antimicrobial efficacy/ safety Antimicrobial agents have been encapsulated in liposomes for two reasons. First, they protect the entrapped drug against enzymatic degradation. For instance, the penicillins and cephalosporins are sensitive to the degradative action of β-lactamase, which is produced by certain microorganisms. Secondly, the lipid nature of the vesicles promotes enhanced cellular uptake of the antibiotics into the microorganisms, thus reducing the effective dose and the incidence of toxicity as exemplified by the liposomal formulation of amphotericin B.70
Constraints in the commercialisation of liposomal preparation Two major constraints are identifiable. First, the lipids needed for their preparation are very scarce, should be of high purity and, hence, expensive. Secondly, liposomal preparations are inherently unstable and require special storage conditions (below 00C) even when the products are freeze-dried. As a result of this stability problem the dosage forms are limited to injection (freeze-dried) powders for reconstitution immediately before use. As a result of these constraints only a few products (e.g., liposomal amphotericin B) have actually been commercialised in spite of the large volume of research on liposomes.
CONCLUSION This review showed that liposomes have been prepared from a variety of synthetic and naturally occurring phospholipids and generally contain cholesterol as membrane stabiliser. Several methods of preparing liposomes were identified, which could influence the particle structure, degree of drug entrapment and leakage of the liposomes. It was also identified that there are improved pharmacokinetic properties with liposomal drugs compared to the free drugs. Further-more, liposomes are tools for drug targeting in certain biomedical situations (e.g., cancer) and for reducing the incidence of dose-related drug toxicity. Instability of the preparations (particularly leakage) is a problem, which is yet to be overcome before full commer-cialisation of the process can be realised. References
© CMS UNIBEN JMBR The following images related to this document are available:Photo images[jm05002f3.jpg] [jm05002f2.jpg] [jm05002f1.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}