 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Medicine and Biomedical Research, Vol. 4, No. 1, June 2005, pp. 37-43 Beta thalassaemia traits in Nigerian patients with sickle cell anaemia CE Omoti Dr C. E. Omoti, Department of Haematology, University of Benin Teaching Hospital, P.M.B. 1111, Benin City, Nigeria. E-mail: ediomoti@yahoo.com; Tel: 2348056014028 Code Number: jm05005 ABSTRACT Haematological values were determined in 246 sickle cell anaemia (SCA) patients in three centres in Benin City, Nigeria, as well as 84 control subjects with haemoglobin A (HbAA) confirmed by haemoglobin electrophoresis at pH 8.6. Automated Coulter counter was used to determine the complete blood counts while the foetal haemoglobin was estimated by the modified Betke method, and haemoglobin A2 by the HbS-free microcolumn chromatography. Six patients (2.4%), comprising of two males and four females, out of the 246 SCA patients were found to have elevated haemoglobin A2 (>3.5%). All the six patients also had elevated haemoglobin F (>1.5%). The family members of three of these six patients were successfully screened. These three patients (1.2%) were found to have positive co-inheritance of thalassaemia trait and sickle cell anaemia. The erythrocyte indices were all reduced in these selected families except for one family whose mean cell haemoglobin concentration was within normal range. Peripheral blood film revealed the presence of target cells and occasional microcytes apart from the sickled cells. The possibility of co-inheritance of the beta thalassaemia gene with the sickle cell gene occurs in about 1.2% of Nigerians with sickle cell anaemia. KEY WORDS: Co-inheritance, anaemia, beta thalassea- mia INTRODUCTION The incidence and distribution, clinical, haematological and pathological features of haemoglobinopathies in West Africa have been amply investigated.1 Haemoglobino-pathies are disorders resulting from inherited abnormalities of globin genes leading to synthesis of a variant haemoglobin from gene mutation, deletion, etc. They are broadly divided into two types, namely, structural haemoglobin variants and the thalassaemia syndromes. The sickle cell disease (SCD), a structural haemoglobin variant, is the most common and most important, accounting for over 60% of the world's major haemoglobinopathies and an estimated 2-3 million individuals are affected in Nigeria.2 The thalassaemia syndromes are a group of inherited disorders in which there is absence or reduced rate of synthesis of one of the globin chains: either the alpha or the beta-globin chains of haemoglobin A (α2β2), which is the major human adult haemoglobin of approximately 97%. The other minor adult haemoglobins are haemoglobin A2 (α2δ2) comprising 2.5% and fetal haemoglobin (α2γ2) comprising less than 1% of the total haemoglobins. The involve-ment of either globin chains causes imbalanced globin chain production, ineffective erythropoiesis, haemolysis and a variable degree of anaemia. The alpha (α) thalassaemia trait, usually due to gene deletion, is caused by loss of one (-α or two (--) of the normal complement of four alpha genes (αα/αα) leading to a decrease in or absence of a chains. Often, it is a presump-tive diagnosis only when no other explanation can be found for microcytic red cells, as it requires sophisticated DNA analysis such as allele-specific oligonucleotide hybridization and polymerase chain reaction (PCR).3 The beta (β) thalassaemia trait, usually due to a point mutation, may be beta (o) or beta (+). A βo thalassaemia is one in which there is no b gene expression and, therefore, no β chain synthesis and consequently no haemoglobin A (HbA) production. A β+ thalassaemia is one in which the β gene is expressed but at a reduced rate so that there is some β chain synthesis and some production of HbA. A patient with â thalassaemia trait will therefore have increased synthesis of haemoglobin F (HbF) and haemoglobin A2 (HbA2) in compensation for the reduced levels of HbA. Hence, diagnosis can be made based upon conducting a positive screening exercise of family members and by the specific laboratory findings, which include increased proportion of HbA2>3.5%, HbF>1.5%, with or without reduced red cell indices,4 all of which will be determined in this study. Since the structural haemoglobin variant and the thalassaemia syndrome occur with a high frequency in some populations, the two types of genetic defect may be found in the same individual.5 This study was designed to find out if there is possibility of a co-existence of beta thalassaemia trait with SCA in a Nigerian population. It was conducted by reviewing the level of HbA2 adjudged to be within the thalassaemic range in SCA patients and to compare this value with the erythrocyte indices that are particularly useful in screening for heterozygous carriers of the thalassaemia gene in population surveys. MATERIALS AND METHODS Patients Two hundred and forty six patients with a diagnosis of SCA regularly attending the consultant outpatient haematology clinic at the University of Benin Teaching Hospital, Central Hospital and the Sickle Cell Centre in Benin City, Nigeria, between August 2001 and July 2002 were recruited into the study after obtaining informed consent. The patients had been diagnosed by demonstration of positive sickling phenomenon and haemog-lobin electrophoresis at pH 8.6 and about whom large numbers of haematological indices and clinical data are available. Family members of the SCA patients were also recruited into the study after consent was obtained. The haemoglobin electrophoretic strip revealed bands on haemoglobins A, F, S and A2 after the haemoglobin S has been confirmed on the selected families. Full clinical examination on each patient was done to rule out any abnormality especially organomegaly. Peripheral blood film was prepared for each patient. The 84 age and sex matched control subjects were made up of primary and secondary school students, medical students and staff of the University of Benin Teaching Hospital. These subjects were free from acute or chronic disease process with no evidence of genetically determined haematological disorder and had HbA on cellulose acetate electrophoresis. Patients were excluded if they had any disorders, such as renal disease, that could affect the haematological values. Sample Collection Five millilitres of venous blood was collected by clean venepuncture from each patient via the antecubital vein using a plastic syringe with minimum stasis, into commercially prepared concentrations of sequestrene ethylene di-amine tetracetic acid (EDTA) bottles. Each sample was then mixed gently and thoroughly to ensure anticoagulation and to prevent cell lysis. An aliquot was used to determine the complete blood counts (CBC) within two hours of collection while the remainder was used to prepare haemolysate for haemoglobin A2 (HbA2) and haemoglobin F (HbF) estimation. METHODOLOGY Complete blood counts (CBC) were analysed using the automated Coulter® ACT diffTM analyser (1997 model). The CBC includes haemoglobin, haematocrit (Hct), red blood cell count (RBCC), mean cell volume (MCV), mean cell haemoglobin (MCH) and mean cell haemoglobin concentration (MCHC). Measurement of HbA2 was done using the chromatographic-spectrophotometric ion exchange-HbS interference free technique using the Biosystem® kit. The procedure recommended by the manufacturer was applied. HbA2 percentage was calculated using the formula: %HbA2 = A415 HbA2 A0 = Total haemoglobin
Measurement of HbF was done using the modified Betke method6 because it is reliable and designed for quantification of small levels of HbF (below 10-15%). HbF percentage was calculated using the formula: % HbF = A413 filtrate x100
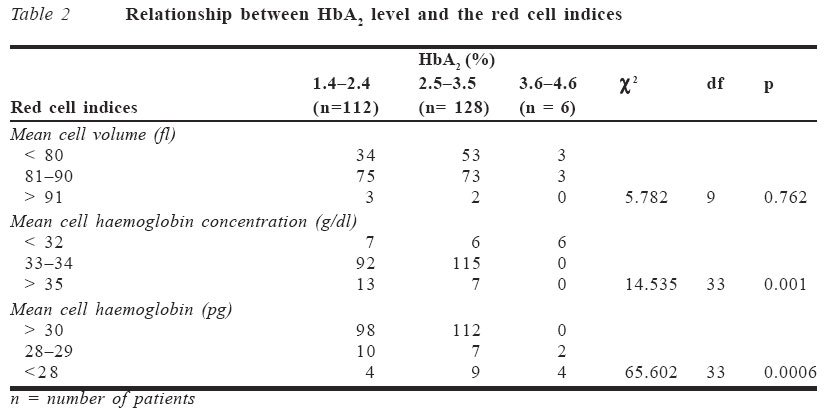
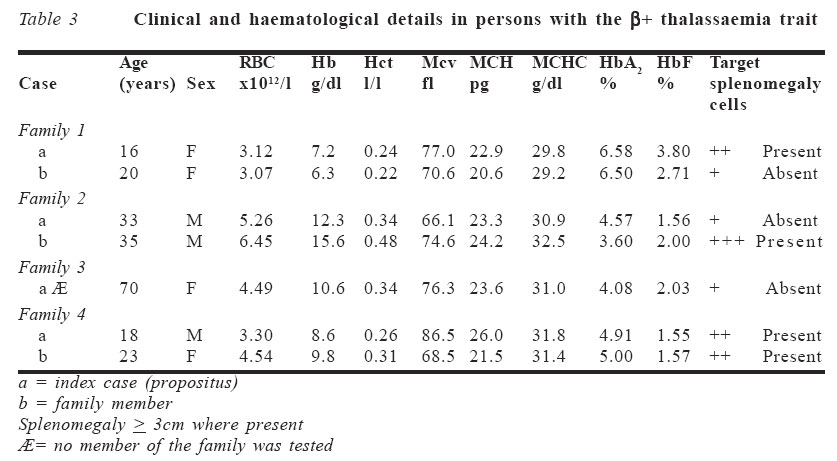
A peripheral blood film was prepared using Leishman's stain, according to Dacie and Lewis method.6 This was to determine the morphological abnormality in the red cells. RESULTS Two hundred and forty six patients, made up of 116 males (47.2%) and 130 females (52.8%), with a diagnosis of SCA, who met the inclusion criteria, were recruited. Their mean age was 23.69 years (S.D ± 10.94) with a range from 5 to 70 years. The haemoglobin A2 (HbA2) level obtained in relation to the sex distribution is shown in Table 1. A total of six patients (2.4%), comprising two males (33.3%) and four females (66.7%), had elevated haemoglobin A2 level above 3.5%. The mean control value of HbA2 was 2.13% ± 0.98, which was significantly lower than in SCA patients (p < 0.01). The mean haemoglobin F (HbF) level obtained in this study was 2.17 ± 1.81. All the six patients with elevated HbA2 (>3.5%) also had elevated HbF (>1.5%). The mean control value of HbF was 1.28 ± 1.04, which was statistically lower than in SCA patients (p < 0.01). The mean cell volume (MCV), mean cell haemoglobin (MCH) and mean cell haemo-globin concentration (MCHC) in control subjects were 84.46fl ± 5.26, 30.16pg ± 2.99 and 34.73g/dl ± 2.76 respectively. They were all significantly higher in controls than in SCA patients (p < 0.01). The relationship between HbA2 level and red cell indices is shown in Table 2. Six patients (2.4%) had high HbA2 levels and reduced red cell indices. Their family members were screened and in three cases (1.2%), one family member each had SCA, high HbA2 levels and reduced red cell indices as shown in Table 3. The red blood cell counts were within normal range for age and sex while only four patients had haematocrit level below 0.31l/l. All the affected individuals with their family members had reduced MCV and MCH, except for the MCHC, which was occasionally overlapping within the normal range. DISCUSSION Little is known about the occurrence and manifestations of thalassaemia trait in Nigeria because it is a rare condition and its diagnosis is considerably more difficult especially in heterozygote subjects. It has also been shown from previous studies that the incidence of beta thalassaemia gene in Africa has been variable.7 This could be because results from previous studies have used either only HbA2 level or erythrocyte indices alone to determine the carriers of the thalassaemic gene, which is subject to underestimation. Esan1 reported an incidence of 0.8% in an indigenous population in western Nigeria while Watson-Williams8 reported an incidence of 0.03% in subjects who had only positive sickling tests. The sickle cell-βo thalassaemia (Sβothal) interaction is characterised by a clinical disorder that is very similar to SCA.9 On the other hand, the interaction of the sickle cell gene with mild forms of β+ thalassaemia (Sβ+thal) may be quite innocuous when compared to patients with haemoglobin S (α2β2s). It is characterised by mild anaemia with haemoglobin concentration of appro-ximately 30% haemoglobin S (HbS), 25%HbA with an elevated level of haemoglobin A2 (>3.5%).5 Thus, a higher than normal HbA2 level in patients with HbS, amongst other parameters, is to be considered before a diagnosis of SCD with thalassaemia is made. However, there is paucity of information regarding evidence for the existence of alpha thalassaemia co-inheritance with sickle cell gene obtained in Nigeria since it requires sophisticated DNA analysis. Despite advances in DNA-based methods, traditional ways of detecting these red cell disorders are still in use because of lack of facilities. The important diagnostic blood counts that can be used in beta thalassaemia trait include increased red blood cell count, reduced erythrocyte indices with some indices overlapping within the normal range, elevated HbA2 and HbF levels, haemoglobin electrophoresis and isoelectric focusing (IEF), which remain useful and point towards more definitive testing if it is warranted.3 All these laboratory tests, including a family study, were determined in the present study apart from the IEF. The prime current indication for DNA-based globin gene analysis is for prenatal diagnosis.3 A secondary indication is for the resolution of diagnostic problems. Elevation of HbA2 above 3.5% was found in six (2.4%) out of 246 SCA patients. However, it was only possible in three cases to evaluate other members of the family where it was found that at least one member also had elevated HbA2, indicating that it was genetically determined.1 Of the remaining three patients, two were lost to follow-up while the elder sister of the third patient, who was also a sickler, had died at a very old age. This third patient was 70 years old with elevated HbA2 and HbF with all the erythrocyte indices reduced. Her late sister probably also had elevated HbA2 and HbF, which may account for the high life expectancy in sicklers in this family. However, excluding this patient and the two lost to follow-up, the frequency of beta thalassaemia trait was estimated to be about 1.2%. This is similar to a Jamaican study that reported a prevalence of 1.5% detected during screening of cord blood.10 Elevation of HbF above 1.5% was also found in all the six patients (2.4%), with all other members of the family also having elevated HbF except in one case (family 3), thus indicating its genetic inheritance. Previous studies have shown that HbF is generally low in Nigerian patients with SCA.11,12 This is in agreement with the low HbF value obtained in this index study. The mean HbF found in this study in SCA and in controls were 2.17 ± 1.81 and 1.28 ± 1.04 respectively. Although the HbF is reported to be low in SCA patients, it was however high when compared to the controls. In contrast to this, other authors have reported mean HbF values of 6.7-9.5%.13,14 This is probably because many of these studies had a high proportion of paediatric SCA patients as well as inherent genetic factor. The latter is said to be responsible for the high HbF in this group of patients. However, HbF levels show significant variations in health and disease states. The estimate given here of 1.2% beta thalassaemia trait in the population of SCA patients was based on the observation of elevated HbA2 and HbF levels, reduced erythrocyte indices and successful family study of three cases. The high incidence reported in other countries, like in the Mediterranean region, may represent differing genetic make-up and reflect selection and varying criteria for the diagnosis, e.g., survey depending on estimations of HbA2 or on MCV alone, thereby overestimating the prevalence of the beta thalassaemia gene as reported in those studies.10 The erythrocyte indices are of consid-erable clinical importance and are widely used in the classification of anaemia. They are usually used in screening programmes for the detection of carriers of β+ thalassaemia in population surveys. Individual values for MCV, MCH and MCHC occasionally overlapped with those in the normal population, thereby casting doubt on the adequacy of these criteria alone in identifying all cases of the heterozygous β+ thalassaemia in SCA patients.10 In this study three of the index patients with elevated HbA2 had reduced MCV of <80fl while the remaining three were within the normal range. All the patients with elevated HbA2 had reduced MCHC of <32g/dl while four of the patients had reduced MCH of <28pg, and the remaining two were within normal range. This implies that these criteria cannot always be used alone to identify the â+ thalassaemia trait. However, the reduced red cell indices and elevated HbA2 level are in keeping with a positive screening programme for the β+ thalassaemia gene. In previous studies patients with low MCV values were observed to also have low values of MCHC and high values of HbA2.15 The HbA2 improves the clinical course of the SCA patients since it is known to reduce the minimum gelling concentration of HbS16 but only at considerably greater concentration of HbA2 than the mean elevation of 0.75% as observed in some studies.15 The clinical and haematological expression of all the index cases and their family members detected during this study were mild. Only three had haematocrit levels below 0.31l/l and all the patients had fetal haemoglobin level greater than 1.50% when compared to the mean control value of 1.28%. The peripheral blood film, apart from the sickled cells, revealed the presence of occasional to numerous target cells (+ to 4+) with few microcytes. These microcytes found in heterozygous thalassaemia are differen-tiated from iron deficiency anaemia and other disorders causing microcytic hypochromic anaemias, e.g., sideroblastic anaemia or certain thalassaemia syndromes. Iron deficiency anaemia is often the first condition considered by a physician because it is the most common after nutritional deficiency anaemia has been ruled out. Although specific laboratory tests to rule out iron deficiency anaemia were not carried out, previous studies have reported a mild erythrocytosis (high RBCC) as characteristic of thalassaemia trait, which agrees with the finding in this study. This is in contrast to findings in iron deficiency anaemia.17 Also, decreased levels of HbA2 are seen in iron deficiency anaemia in contrast to increased HbA2 level in S-beta thalassaemia as seen in this study. No clinical feature of iron deficiency anaemia was present. Splenomegaly persisted significantly in four patients. This is compatible with decreased intravascular sickling and preservation of the splenic capillary.15 The persistent splenomegaly in the disease has been found to correlate with high levels of HbF as shown in this study. None of these patients had any need for blood transfusion in the preceding year, as none was anaemic. In conclusion, there is likely a coexistence of beta thalassaemia trait in patients with SCA in Nigeria. These patients have high HbA2 and HbF levels, reduced erythrocyte indices and positive family history suggesting that this is a heritable trait. References
© CMS UNIBEN JMBR The following images related to this document are available:Photo images[jm05005t3.jpg] [jm05005t2.jpg] [jm05005t1.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}