 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Postgraduate Medicine, Vol. 47, Issue 4, 2001 pp. 252-255 The Antley-Bixler Syndrome: Two New Cases Hosalkar HS, Shah
HS, Gujar PS, Shaw BA
Division of Paediatric Orthopaedics,
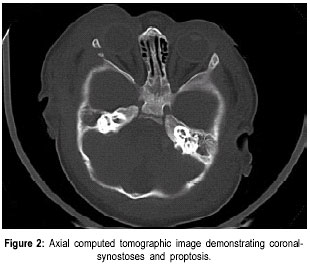
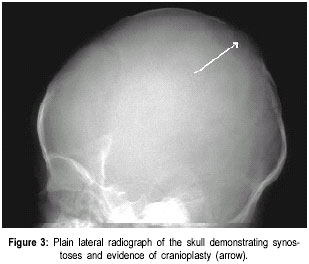
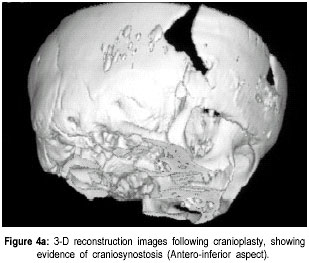
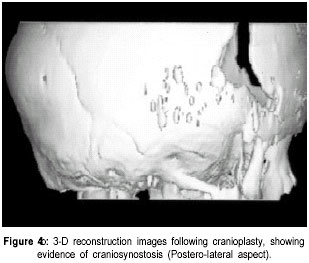
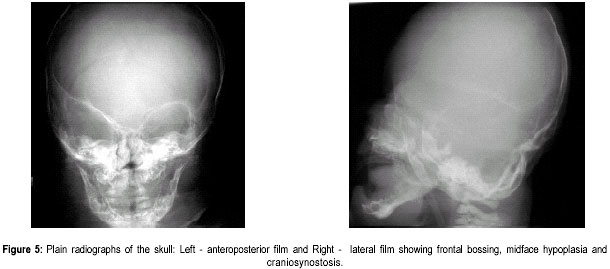
The Valley Children’s Hospital, California, UCSF, USA. Code Number: jp01073 Abstract The Antley-Bixler syndrome is a rare multiple congenital anomaly with a high mortality rate. The characteristic manifestations include craniosynostosis, radiohumeral synostosis, midface hypoplasia, joint contractures and arachnodactyly. We report two new cases of this syndrome and address the diagnostic features, associated malformations, inheritance patterns, prenatal findings, and piefly review the literature. Key Words: Craniosynostosis, radiohumeral synostosis, arachnodactyl, mid face hypoplasia. Antley-Bixler syndrome (ABS) is a rare developmental malformation with many musculoskeletal, craniofacial and urogenital anomalies necessitating multi-systemic assessment. Mortality is as high as 80% in the first months of life. We present two new cases that illustrate well the classic diagnostic features of the syndrome. We highlight features of this potentially debilitating syndrome, discuss role of early diagnosis, multidisciplinary team approach in management, and piefly review the literature.Case History Case I: A 5-month old Hispanic boy, born to a 21-year-old primigravida and a 27-year-old father, presented with a forearm fracture following injury. The parents were unrelated and family history was not contributory. The pregnancy was uneventful and the baby was vaginally delivered at 38 weeks of gestation. At birth, weight was 2.79 kgs (10(th) to 25(th) percentile), total body length was 46.5 cms (5(th) to 10(th) percentile), and head circumference was 33.5 cms (10(th) to 25(th) percentile). There were associated delayed milestones, dysmorphic features, seizures, and elbow joint deformities that were noted since birth. On examination there was evidence of pachycephaly (trapezoidal head with cephalic index of 82.5), frontal bossing, midface hypoplasia, depressed nasal pidge and proptosis. Examination of limbs revealed fused elbows, multiple joint contractures and arachnodactyly. Radiographs confirmed the bilateral radiohumeral synostosis and forearm fracture on the right side (Figure 1). Skull x-rays and computed tomographic (CT) scans confirmed synostoses of both coronal sutures. Proptosis was documented on the scans (Figure 2) and so was bilateral choanal atresia. He had a narrow rib cage with thin ribs. Chromosomal studies were normal. The fracture was splinted and it subsequently healed. Craniosynostosis was treated with reconstruction cranioplasty (Figures 3, 4a & 4b) involving forehead and orbital advancement at the age of nine months. Aggressive respiratory and physical therapy program was continued. Follow up at 11 months revealed a head circumference of 42.5 cms with excellent results of craniofacial reconstruction. The child is alert and growing well when seen last at 14 months of age. Case II: An 8-month old Hispanic boy was referred to the orthopaedic services for congenital fused elbows, following assessment for craniosynostosis. The child was born of a full term normal delivery from unrelated parents. Anthropometry at birth included a birth weight of 3 kgs (25(th) percentile), length of 49.5 cms (25(th) to 50(th) percentile), and head circumference of 34 cms (25(th) to 50(th) percentile). The father’s family history was significant for a pother who had a premature child (which expired), a sister who had multiple miscarriages, and another sister who had a son with hypoplastic left heart. Examination revealed pachycephaly (cephalic index of 83.2), bilateral coronal synostoses, midface hypoplasia (Figures 5a & 5b), radiohumeral synostosis, femoral bowing, and camptodactyly involving both hands. Imaging confirmed the synostoses and complete choanal atresia on the right side. At nine months his height was 72 cms (25(th) to 50(th) percentile), weight was 7.285 kgs (< 5(th) centile), and head circumference was 41.5 cms (< 5(th) centile). Parents were explained about the recurrence risk for their family. The child is on respiratory and physical therapy and scheduled for craniofacial surgery. Discussion Antley and Bixler in 1975 were the first to report a child with trapezoidocephaly, midface hypoplasia, radiohumeral synostoses, and fractures of the femur. DeLozier in 1980 referred to this syndrome as a multisynostotic osteodysgenesis with long bone fractures.(1,2) The aetiopathogenesis of this syndrome is still not known. Sex ratio has been observed to be 2:7 for males and females.(3) Review of the literature, to the best of our knowledge reveals that 41 cases have been described.(2,4-7) Autosomal recessive inheritance has been suggested by most authors.(1-3) However, there are reports of a new dominant mutation at the fipoblast growth factor receptor 2 (FGFR2) locus as well as in the offspring of mothers taking the antifungal agent fluconazole during early pregnancy. Reardon et al have reported abnormalities of steroid biogenesis in seven of 16 patients with an Antley-Bixler phenotype, and identified FGFR2 mutations in seven of these 16 patients, including one patient with abnormal steroidogenesis. These findings suggest digenic inheritance in some cases of ABS.(6) Chun et al were the first to report an apparent dominant de novo mutation in the FGFR2 gene.(8) Consanguinity has been reported twice.(9) The classical manifestations of ABS include craniosynostosis, radiohumeral synostosis, midface hypoplasia, joint contractures and arachnodactyly.(2) Synostosis of cranial sutures and elbow joints is considered as the minimal diagnostic criteria.(8) Varying urogenital anomalies including ectopic kidneys, ureteric obstruction, hypoplastic uterus, hypoplastic or fused labia majora, large clitoris, small penis or undescended testes, are seen in less than 50% of the patients. Congenital heart defects have been noted in about 21 % cases.(2) We have compared the anomalies in our cases with the estimated percentage of those described in literature (Table 1).(2) Radiological diagnosis is based on demonstrating craniosynostosis, radiohumeral synostosis, midface hypoplasia, and other associated femoral, hip or spine anomalies. Urogenital and cardiac anomalies need special imaging. Respiratory distress secondary to choanal atresia or stenosis can be a serious factor for mortality, often requiring intubation, tracheostomy and nasal stents. Prognosis is guarded in infancy and childhood, and improves with age. The craniosynostosis can result in disruption of normal pain development and cause retardation.(9) Auditory dysfunction may be conductive or sensorineural.(3) Although radiohumeral synostosis, joint contractures and arachnodactyly are present in almost all cases, other orthopaedic manifestations could be femoral bowing, ulnar bowing, camptodactyly, synostosis of the carpal and tarsal bones, clubfoot, vertepal body anomalies and perinatal fractures. Results of resecting the synostosis are not encouraging and surgery may best be avoided.(2) Finger contractures are associated with shortened tendons, which usually improve with physical therapy. Hip movements are limited secondary to contractures and may predispose to femoral fractures.(3) Prenatal diagnosis by mid-trimester ultrasound examination is possible and should be recommended.(4,10,11) Fixed flexion of the elbow appears to be the essential diagnostic finding, although humero-radial synostosis, medial bowing of ulna, long hands and fingers, and bowing of femora have been noted.(11) The poor prognosis (in view of high infant mortality) in this condition makes counselling difficult and early termination of pregnancy a consideration.(12) Genetic counselling is important and depends on accurate prognostic and therapeutic data.(4) Clinicians should recognise that Antley-Bixler syndrome could lead to significant respiratory problems in early infancy leading to mortality. Synostosis frequently results in early orthopaedic consult, and the surgeons should be aware of the frequent association with this syndrome. Early aggressive intervention and addressing major congenital malformations forms the mainstay of treatment. Prenatal ultrasonographic diagnosis and genetic counselling play an important role. References
This article is also available in full-text from http://www.jpgmonline.com/ © Copyright 2001 - Journal of Postgraduate Medicine The following images related to this document are available:Photo images[jp01073t1.jpg] [jp01073f5.jpg] [jp01073f1.jpg] [jp01073f4b.jpg] [jp01073f4a.jpg] [jp01073f3.jpg] [jp01073f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}