 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Postgraduate Medicine, Vol. 48, Issue 1, 2002 pp. 50-51 Robinow syndrome Hosalkar HS, Gerardi J, Shaw BA Division of Paediatric Orthopaedics, The Valley Children's Hospital, Fresno,
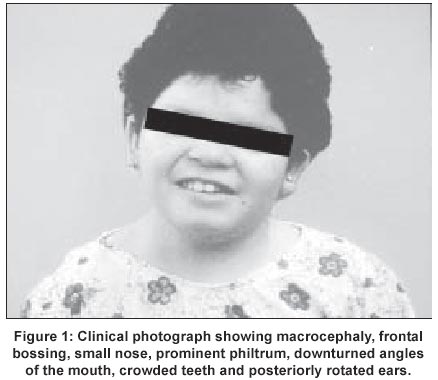
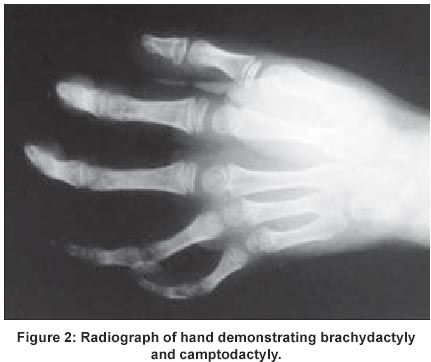
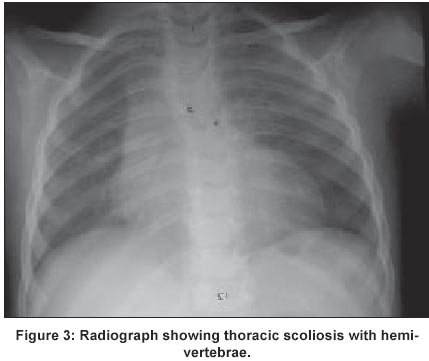
UCSF, USA Code Number: jp02015 A 10-year Hispanic girl, born of unrelated parents (first child of a 36-year-old woman and a 42-year-old man), after a normal pregnancy, had presented with short stature and facial dysmorphism. There was no significant peri-natal history and family history was negative for birth defects and genetic disorders. Anthropometry at 6-months revealed weight of 4300 gms, height of 54 cms, and head circumference of 36 cms (all < 3rd centile). There was history of neonatal jaundice and recurrent respiratory infections in infancy and childhood. Examination revealed dysmorphic facial features (including macrocephaly, frontal bossing, hypertelorism (interpupillary distance of 7 cms), prominent eyes, down-slanting palpebral fissures, small nose, prominent philtrum, down turned angles of the mouth, crowded teeth and posteriorly rotated ears) (Figure 1), brachymelia with brachydactyly, camptodactyly (Figure 2), mid-thoracic scoliosis with hemivertebrae and rib crowding (Figure 3), and genital anomalies in the form of hypoplastic clitoris and labia majora. She had hypoplastic nails in all digits, hyperlaxity of joints in upper and lower limbs, with associated skin laxity. There was no evidence of any umbilical or abdominal wall anomalies and no associated organomegaly. She had a cleft palate that was repaired at the age of 2 years. Anthropometry (at age 10) revealed a height of 136.5 cms (25th-50th percentile), weight of 39.5 kg (75th-90th percentile), mesomelic dwarfism with UL: LL ratio of 1.6 and forearm brachymelia with arm to forearm ratio of 1.5: 1. She was Tanner stage I in terms of sexual maturity, had obvious neurological and developmental delay with a functioning I.Q at a 4-year-old level. Her bone age was 8 years 5 months at a chronologic age of 10 years. CT brain revealed an absent corpus callosum, while cardiac assessment did not reveal any associated anomalies. Endocrine work-up (Growth hormone, LH, FSH, oestrogen, progesterone, IGF-1, T3,T4, and, TSH) as well as immunological work up (T-cell and B-cell functions) was normal. A diagnosis of Robinow syndrome was made.
Discussion First described by Robinow et al in 19691 this syndrome refers to a combination of short stature, characteristic facial dysmorphism (foetal facies), genital hypoplasia, and mesomelic brachymelia. Although the incidence of Robinow syndrome is about 1:500,000, the prevalence is slightly lower since 5-10 % of patients die in infancy or early childhood. The male to female ratio of Robinow syndrome is 1:1.2 The essential criteria required for clinical diagnosis are characteristic facies, forearm brachymelia, short stature, and genital hypoplasia.2 Autosomal dominant, autosomal recessive, and male-to-male transmission have all been reported in this syndrome, thus suggesting genetic heterogeneity.3 Phenotypic presentation is related to the inheritance pattern and patients with the recessive form have more severe dwarfism, more severe vertebral anomalies, more severe brachymelia and more digital defects.2 Parental age has not been found to be a significant factor although consanguinity seems to play some role.3 Chromosome studies have revealed consistent negative results except for a single reported case of 7q (7q32®q ter) deletion.4 Mutation of the gene encoding the ROR2 tyrosine kinase has now been implicated in Robinow syndrome.5,6 No specific metabolic abnormalities have been identified. Lee et al observed partial primary hypogonadism as indicated by elevated levels of follicular stimulating hormone, hyper-response of LH on stimulation, normal 5 alpha-reductase activity and normal androgen receptor activity in genital skin fibroblasts.7 Radiographs are useful for assessing spinal deformities or rib anomalies and may also help to document forearm brachymelia, hand anomalies, and facial features. Assessment of the length of the long bones and the ulna / humerus ratio has been reported a useful index for the prenatal diagnosis.8 Pulmonary or cardiac complications are common in these patients, and approximately 5-10% have a premature death in infancy and childhood.1,2 Although some degree of mental retardation occurs in few (20%), intelligence is normal in most cases.3 Management includes surgical intervention for hernias, bracing or surgery for vertebral anomalies and scoliosis, orthodontic treatment for dental malalignment, facial reconstruction in selected cases for psychological support, and occasionally hormonal therapy. Recombinant human growth hormone has been proved to cause a significant increase in the growth rate in children with Robinow syndrome and growth hormone deficiency.9 Human chorionic gonadotropin and/ or testosterone therapy during infancy has been suggested to improve penile length and testicular volume in male children.10 With normal intelligence in most cases, and adequate sexual functioning and reproduction,2 the prognosis for Robinow syndrome is good.
References
This article is also available in full-text from http://www.jpgmonline.com/ © Copyright 2002 - Journal of Postgraduate Medicine The following images related to this document are available:Photo images[jp02015f2.jpg] [jp02015f3.jpg] [jp02015f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}