 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
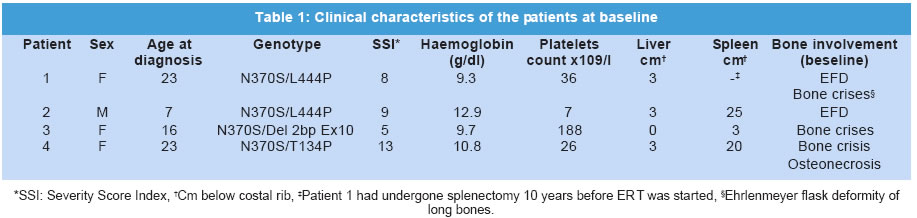
Journal of Postgraduate Medicine, Vol. 49, No. 2, April-June, 2003, pp. 127-131 Brief Report Extended Interval between Enzyme Therapy Infusions for Adult Patients with Gaucher's Disease Type 1Pérez-Calvo J, Giraldo P,1 Pastores GM,2 Fernández-Galán M,3 Martín-Nuñez G,3 Pocoví M4 Department of Internal Medicine. Hospital Clínico Universitario "Lozano
Blesa", Zaragoza, Spain,
1Department of Hematology. Hospital Universitario "Miguel Servet",
Zaragoza; Spain,
2Neurogenetics Program. New York University School of Medicine.
New York; USA, 3Department of Hematology. Hospital "Virgen del
Puerto", Plasencia (Cáceres); Spain and
4Department of Biochemistry and Molecular Biology. University of Zaragoza;
Spain
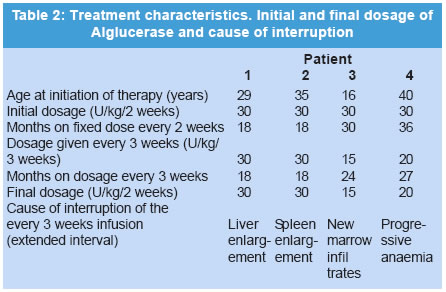
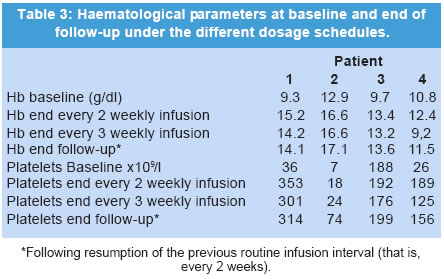
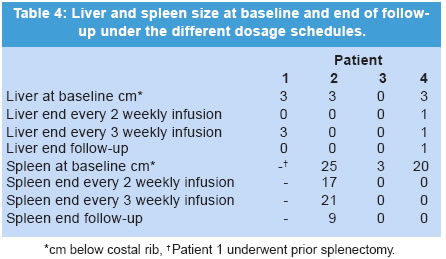
The manuscript was presented as part at 5th Workshop of the European Working Group on Gaucher Disease. Prague, Czech Republic, on May 4, 2002. Code Number: jp03034 Abstract: BACKGROUND: Enzyme replacement therapy (ERT) for Gaucher's disease with alglucerase or imiglucerase is efficacious, well-tolerated and safe. However, cost considerations, visits to medical facilities, potentially duration of theray for life, are issues of major concern to a proportion of treated patients and has, in some cases, led to the withdrawal of therapy. AIMS: To elucidate whether an extension of the interval between enzyme infusions to once every three weeks is as effective in maintaining the clinical responses achieved with the bi-monthly regimen. MATERIALS AND METHODS: Four patients with an optimal response to ERT (at 30 units/kg every two weeks for an average of 27 months), were subjected to enzyme dose/frequency changes that essentially constituted a reduction in cumulative dose over the treatment period. Patients were assessed every 6 months for alterations in haematological parameters, plasma chitotriosidase levels, liver and spleen size, and bone symptoms. RESULTS: All patients had to resume the previous infusion schedule of once every two weeks; one because of new bone marrow infiltrates, two because of visceral enlargement, and the fourth due to progressive anaemia. CONCLUSIONS: This limited experience suggests that a reduction in enzyme dose associated with an extended interval between infusions may lead to variable disease control, and underscores the need for individualization of enzyme therapy. (J Postgrad Med 2003;49:127-131) Key Words: Enzyme replacement therapy, Gaucher's disease, alglucerase, imiglucerase Over a decade ago the first two reports were published on the treatment of Gaucher's disease (GD) patients through intravenous supplementation of the deficient enzyme, acid b-glucocerebrosidase (OMIM *606463).1,2 Since then, it has been clearly shown that enzyme replacement therapy (ERT), with either the placental-derived alglucerase (Ceredase®), or the recombinant formulation imiglucerase (Cerezyme®), is an efficacious, well-tolerated and safe treatment modality. However, there are several important issues associated with the current treatment regimen that pose difficulties in administering ERT to all patients. The cost of the drug is much higher than that of most drugs currently used in other clinical situations. In addition, repetitive injections necessitate patients to attend medical facilities periodically, thereby limiting their independence, and in some cases, their life-style. In a recent report, the costs and the required commitment to regular, frequent intravenous infusions were cited as the two main reasons behind the withdrawal of ERT.3 A less frequent administration of the enzyme, i.e. one infusion every 3 or 4 weeks, as suggested by Brady in 1994,4 has the potential benefit of simultaneously reducing the cost and improving quality of life. However, to date no studies on the efficacy of such a schedule have been reported. In the present report, we discuss the results of the follow-up of a group of four adult GD patients treated with a conventional ERT schedule (once every 2 weeks), followed by a less frequent regimen, consisting of one infusion every 3 weeks. Patients and Methods The clinical characteristics of the patients, all diagnosed with type 1 GD, are presented in Table 1. The dosage of 30 U/kg every two weeks is the most commonly used regimen in Spain, given the demonstrated efficacy and lower associated costs.5 The study patients had been on ERT with alglucerase (Ceredase ® Genzyme Therapeutics, Cambridge, MA, USA) at a dose of 30 units/kg every two weeks for an average of 27 (range 18-42) months, and a significant positive clinical and haematological response had been achieved in all of them. In two cases (patients 1 and 2) the dose of alglucerase administered per infusion was unchanged; however, the infusion interval was increased to once every three weeks. In the other 2 patients (patient, 3 and 4), treatment continued to be administered once every two weeks with a reduction in the dose of alglucerase to 15 U/kg/2 weeks and 20 U/kg/2 weeks, respectively. In both patients, at 30 and 36 months respectively, the administration interval was then increased to one infusion every 3 weeks with no changes in the previous amount of alglucerase given per infusion (Table 2). Thus, all patients essentially had reductions in their cumulative enzyme dose during the latter course of treatment. All patients underwent physical examination, routine blood tests, measurement of plasma chitotriosidase level and abdominal ultrasonography every 6 months. Other specific tests (i.e. MRI) were performed as needed. Results The data on clinical, analytical and major parameters that were monitored for each patient under the different dosage schedules are shown in tables 2, 3, 4. After a variable period of time, all patients receiving every-3-week infusions had to resume the former every-2-weeks regimen since some sign/s of deterioration had developed (Table 2). Patient 1: After 18 months of receiving 30 U/kg every 3 weeks her liver regained its initial (pre-treatment) size. Her infusions were then switched back to 30 U/kg every 2 weeks with no further complaints. Patient 2: After 18 months of receiving 30 U/kg every 3 weeks, an increase in spleen size to values similar to those at baseline prompted us to resume the previous regimen of 30 U/kg every 2 weeks. This treatment schedule has been maintained for an additional 4-year period without any further signs of disease progression. Patient 3: This female patient started enzyme infusions at a dose of 30 U/kg of alglucerase every 2 weeks. After the first year of therapy, alglucerase was tapered, at 6 months interval, to a minimum of 15 U/kg every 2 weeks. This schedule was maintained for 30 months. Thereafter, she was given 15 U/kg every 3 weeks for 24 months. No liver or spleen enlargement was detected and her haematological parameters remained stable throughout this period. However, at the end of the second year on an every 3-weekly infusion regimen she complained of mild arthralgia localised in the right knee and both ankles without any signs of inflammation. An MRI of the spine showed a subtle increase in the degree of marrow infiltration within the body of the 4th and 5th lumbar vertebrae, without any changes in the long bones. Her enzyme infusions were increased to 15 U/kg every 2 weeks followed by relief of symptoms. A year later, the MRI was repeated and showed that marrow infiltration of the vertebral bodies had cleared. She has been maintained on this regimen for about 3 and half years without any new findings. Patient 4: This female patient started ERT with 30 U/kg of alglucerase every 2 weeks, and was maintained on this regimen for 3 years. Subsequently, her dose of alglucerase was reduced to 20 U/kg every 2 weeks for one year, followed by a change in infusion frequency to 20 U/kg every 3 weeks. Finally, after 24 months of the extended infusion regimen, a bi-monthly schedule was again implemented due to progressive anaemia. Discussion A decade after the introduction of ERT in GD, it is possible to state that this modality of treatment is efficacious, safe and well tolerated.6,7 However, there are certain issues that remain to be resolved. Whatever regimen is used to start therapy - whether high-dose low-frequency or low-dose high-frequency - the minimum effective dose after an initial response is obtained tends to be about 30 U/kg per month.8,9 This still represents a significant financial burden. A low-dose high-frequency regimen was proposed as an attempt to mitigate the financial burden of ERT without loss of efficacy,10 but this leads to greater interference in the patients' life-styles, which can be so inconvenient that some patients actually prefer to give up therapy. Furthermore, there is heterogeneity in clinical response to therapy and a low-dose regimen may not be adequate to control or reverse disease symptoms.11,12 The fact is that, whether due to financial concerns or to a variety of personal reasons, withdrawal of ERT is a relatively common issue for some patient groups.3 Currently, there are no guidelines for the long-term treatment of patients with GD after they have responded to and have been on ERT for several years. The usual practice is to reduce the total amount of the drug but keeping the dose interval and the rate of infusions ongoing indefinitely. Brady suggested in 1994 that it was conceivable that less frequent injections of the enzyme, i.e. once every 3 to 4 weeks, may be efficacious in patients who had been "normalized" after the initiation of treatment.4 This is an attractive approach since it would reduce the financial burden and lifestyle adjustments associated with the current treatment regimen. Although the recommended changes appear reasonable and the fact that one splenectomized adult patient has been stabilized on single monthly injections,13 this treatment schedule has not been introduced to a larger cohort of patients, and no further data relating to this issue have been reported. In this report, we have explored the efficacy of extending the interval between infusions of alglucerase in GD patients, and have considered either a reduction of the amount of the enzyme given and then subsequently increasing the interval of administration, or alternatively extending the infusion interval without any changes in the amount of alglucerase given per infusion. All of the patients in our study had a protective N370S allele and started treatment after full skeletal growth had been achieved. These patients had previously responded to the conventional ERT schedule and were subsequently kept on an every three weeks infusion regimen for as long as 27 months. Follow up evaluation revealed that all four patients had developed findings suggestive of an active disease process. Two patients had significant visceral enlargement, one developed progressive anaemia and another, presented with abrupt skeletal complications and new vertebral marrow infiltrates that prompted us to return to the often-used schedule of infusions every 2 weeks. Elstein and colleagues reported the clinical course of 15 patients with interruption of therapy after an initial positive response. Although they found no major clinical complications over a variable length of follow-up monitoring, 6 of the patients required the re-institution of treatment because of a clinical relapse.3 In addition, other authors have found that cessation of ERT is usually followed by the recurrence of GD-related symptoms that requires reinstitution of ERT.14,15 The heterogeneity in clinical responses suggests that some especially sensitive (i.e., highly responsive) patients may achieve a sustained response on less frequent infusion schedules. However, it is possible that the amount of enzyme administered after delaying the infusion for an extra week is too low to reach the therapeutic threshold9 necessary to elicit a long-term response. On the other hand, the lack of efficacy could also be explained by the delay of treatment administration irrespective of dose. As a consequence, in our experience, increasing dose interval appears to lack the effectiveness of the commonly used regimen (i.e., an infusion once every two weeks). It is possible that disease control may be effectively maintained during less frequent infusion intervals if the administered dose is increased. However, this issue has not been examined systematically and maintenance on a high dose would not address concerns regarding treatment costs. Larger, prospective controlled studies are still needed to draw broadly applicable guidelines for the optimal management of patients with Gaucher's disease. Acknowledgement This study was supported by a grant from Fondo de Investigaciones Sanitarias Ministerio de Sanidad y Consumo (FIS n0 00/0546) and by the "Fundación Española para el Estudio y Terapéutica de la Enfermedad de Gaucher" (FEETEG). References
Expert's Comments - The need to define guidelines for maintenance enzyme replacement therapy for Gaucher disease: expanded intervals, dose modification, or drug vacations? Enzyme replacement therapy (ERT) has revolutionised the clinical management of patients with Gaucher disease. Although the seminal trial involved only 12 patients for less than a year,1 in the ensuing 13 years, and with experience in more than 3000 patients world-wide, its safety and efficacy have been abundantly documented.2 However, as these patients achieve near-normalisation of disease parameters and, as new patients are diagnosed and treated prior to advent of irreversible signs and symptoms, there is a need to consider establishing guidelines for maintenance and/or withdrawal of therapy after certain clinical criteria have been met. In an attempt to partially address this issue, a clinical trial of switch-over to the oral substrate inhibitor, OGT 918 (Zavesca®, Oxford Glycosciences Ltd), was devised in patients with stable disease parameters after a minimum of two years of enzyme therapy.3 A no-treatment arm was deemed unethical since standard, effective treatment was available. In this latter trial there was no deterioration in any disease parameter in patients who switched to OGT 918 or who took the two therapies in combination during the six months of the trial. Clinical trials too are underway in the US in patients stabilized on enzyme therapy to receive unaltered dosage but with frequency reduced to once every four weeks. In both of the above, signs and symptoms of the disease are the primary endpoints. It has been our reported experience that 14 adult patients who had stopped enzyme treatment (durations from 5-56 months) for various personal reasons (withdrawal periods of 8-47 months) did not revert to respective baselines nor suffer exacerbation of bone or lung involvement.4 Indeed, some patients apparently continued to benefit from enzyme therapy even after withdrawal. At the level of improving the quality of life of patients who remain "chronically ill" by virtue of commitment to unmodified drug regimens rather than because of unimproved or life-threatening disease, reducing the frequency of intravenous infusions is an important step forward. The authors of the current study present four patients who met accepted criteria for enzyme replacement.5 At the end of their respective periods of therapy, with good response in all patients, the frequency of infusions was reduced from once every two weeks to once every three weeks. Primarily on the basis of elevation in levels of activity of the surrogate marker chitotriosidase, the original infusion schedule was re-instituted in each patient; the authors imply that extending the interval of treatment may result in deterioration of disease parameters to baseline values. Leaving aside the financial aspects of expensive therapies in countries with limited resources which is an argument to reduce dosage rather than frequency of administration but is of considerable impact as well, this study is an opportunity to garner support for a consensus report by responsible experts for maintenance protocols. While the authors of this limited series have reached a diametrically opposite conclusion, we applaud their initiative in crediting the need to simplify criteria as well as institute innovative regimens that highlight patient convenience without sacrificing good clinical practice. Nonetheless, it is important to note that an increase in a very sensitive surrogate marker is not necessarily predictive of imminent clinical deterioration. Thus, we would recommend for enzyme-stabilised patients, disease-specific clinical parameters such as symptoms, organ volumes, and/or blood counts as criteria for candidacy for decreased frequency or dosage (or withdrawal) as well as (when appropriate) for re-instatement of enzyme replacement therapy. Ari Zimran, MD and Deborah Elstein, PhD Gaucher Clinic, Shaare Zedek Medical Center, Jerusalem, Israel References
Copyright 2003 - Journal of Postgraduate Medicine. Online full-text also available at http://www.jpgmonline.com/ The following images related to this document are available:Photo images[jp03034t5.jpg] [jp03034t1.jpg] [jp03034t4.jpg] [jp03034t3.jpg] [jp03034t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}