 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
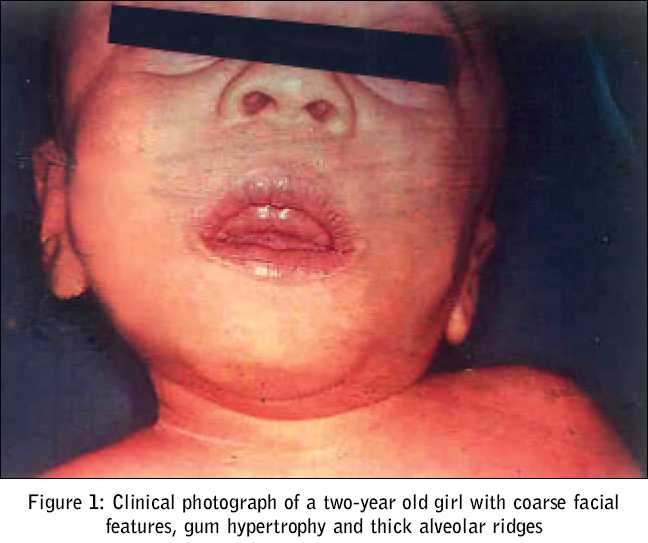
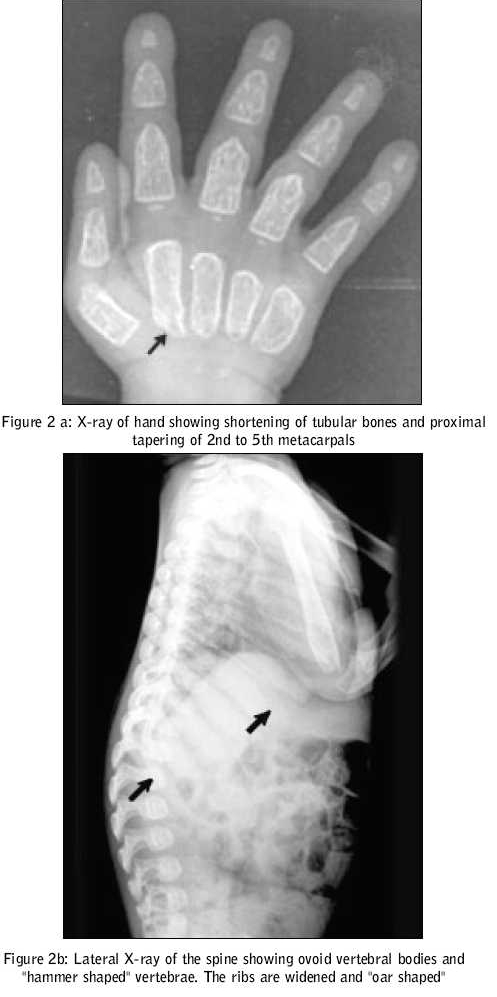
Journal of Postgraduate Medicine, Vol. 51, No. 3, July-September, 2005, pp. 232-233 Images In Medicine Mucolipidosis II (I - Cell Disease) Kumar TS, Scott JX, Raghupathy P, Moses PD Department of Child Health, Christian Medical College, Vellore Code Number: jp05084 A two-year-old girl presented with abnormal facies and delayed development since birth. As she hailed from an orphanage, historical details regarding her birth and family were not known. She had failure to thrive, with her weight being static for the preceding one year. Her developmental milestones had been grossly delayed. She had had two episodes of lower respiratory tract infection and one episode of diarrhoea requiring hospitalisation and administration of intravenous antibiotics. On examination, she was found irritable, but her vital signs were stable. On anthropometry, her length/height was 58 cm (70 percent of expected height), weight 4.7 kg (47 percent of expected), head circumference 40 cm (< 3rd percentile), upper segment to lower segment ratio 1.5: 1, mid-arm circumference 11 cm. She had coarse facial features, a narrow forehead with temporal narrowing, thickened oedematous eyelids, low-set ears with long ear lobules, anteverted nares, flat occiput, depressed nasal bridge, gum hypertrophy, thick alveolar ridges, long philtrum, short neck and thick skin. Her joints were stiff. She had corneal clouding. An examination of the respiratory and cardiovascular systems revealed no abnormality. An abdominal examination revealed a soft hepatomegaly of 4 cm [Figure - 1]. Investigations revealed haemoglobin of 9.1 gm%, total white cell count of 21400/cu.mm, differential count of Lymphocyte 83%, Neutrophils 12%, Monocytes 5%, and platelet count of 5, 01,000/cu.mm. Her serum calcium, inorganic phosphorus, alkaline phosphatase and renal and liver function tests were normal. Bone marrow examination yielded normocellular marrow with erythroid hyperplasia and decreased iron stores. Slit lamp examination showed bilateral corneal clouding. Her 24-hour urine mucopolysaccaride estimation was 0.7 mg, which was within normal limits. Skeletal survey revealed signs of dysostosis multiplex. Lateral X-ray of the spine revealed ovoid vertebral bodies with hypoplasia of anterosuperior corners and "hammer shaped" vertebrae. The ribs were widened and "oar shaped" [Figure - 2]a. An x-ray of the long bones of both lower limbs revealed osteoporosis. There were typical abnormalities in the hands, that is, rectangular and shortened metacarpals with coned proximal ends (2nd to 5th) [Figure - 2]b. She was diagnosed to have Mucolipidosis type II (I cell disease) on the basis of clinical, biochemical and radiological features. Discussion I-cell disease (Mucolipidosis II) is a rare autosomal recessive genetic disorder of lysosomal metabolism. Leroy and De Mars in 1967 distinguished mucopolysaccharidoses and lipidoses.[1] Three distinct entities described by Gilbert et al include a prototype, a malignant infantile and a benign juvenile form.[2] Even though the molecular basis of I- cell disease remains unknown, it was suggested that mutations of different subunits of the N-acetyl-glucosaminyl-phosphotransferase molecule were responsible.[3] These children have psychomotor retardation, short stature and Hurler-like features. In our child these features were apparent from early infancy. Coarse facial features, low-set abnormal ears, high arched palate, thickened eyelids, gingival hypertrophy, corneal clouding, tight skin, brownish hair and hirsuitism are the usual described features. Our patient had obvious gingival hypertrophy at the end of one year of life. In Mucolipidosis II, the abdomen may be protuberant with umbilical or inguinal hernia, and mild to moderate hepatomegaly. Gross motor development is gradually retarded, as the patient gets older. Convulsions are rare. Mental development is noticeably better than motor development[4]. Both mental and motor functions were severely affected in our patient. Children with I-cell disease usually have joint contractures, kyphoscoliosis and gibbus. Chest deformities, congenital dislocation of hip joints and pes valgus may be seen. Systolic murmurs, significant hypertrophy of left ventricular wall and interventricular septum may be seen. In some cases, leaflets of aortic and mitral valves may also be hypertrophic. The radiological abnormalities in mucolipidosis II are those of severe dysostosis multiplex rather like mucopolysaccharidosis I-H (Hurler). In Hurler′s syndrome the radiology becomes characteristic after several months, whereas in I-cell disease, the abnormalities are first observed in the neonatal period itself.[5] The clinical course is progressive and these children usually die by the age of four. I-cell disease is universally a fatal genetic disorder. Prenatal diagnosis can be offered in pregnancies at risk. References
Copyright 2005 - Journal of Postgraduate Medicine The following images related to this document are available:Photo images[jp05084f2.jpg] [jp05084f1.jpg] |
| |||||||||

{kind=link}
{kind=link}