 |
search
for |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
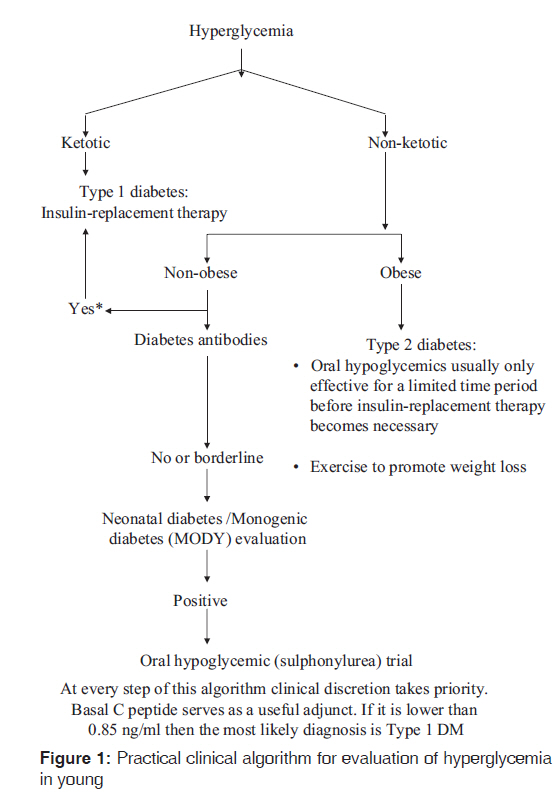
Journal of Postgraduate Medicine, Vol. 57, No. 4, October-December, 2011, pp. 270-271 Guest Editorial Managing non-ketotic childhood hyperglycemia V Shivane, A Lila, T Bandgar, N Shah Department of Endocrinology, Seth G.S. Medical College and K.E.M. Hospital, Mumbai, Maharashtra, India Code Number: jp11080 PMID: 22120853 The prevalence of diabetes mellitus (DM) is increasing worldwide, and DM is not sparing even the children or adolescents. Childhood hyperglycemia is classically divided into Type 1 DM and Type 2 DM. Type 1 diabetes is characterized by cell-mediated autoimmune destruction of the pancreatic beta cells, leading to absolute insulin deficiency. [1] In contrast, Type 2 diabetes is characterized by insulin resistance (secondary to obesity) and variable degrees of insulin deficiency. It is important to recognize that pancreatic beta cell dysfunction is the primary pathophysiology of any form of diabetes, regardless of its "type". In a child, presentation with severe hyperglycemia and ketosis is usually due to Type 1 DM, which can be further confirmed by diabetes-associated antibodies and low serum c-peptide levels. [2],[3] Patients with obesity, acanthosis nigricans and hyperglycemia commonly presents with Type 2 DM. The clinical case need not always be a classical presentation and the physician needs to consider uncommon causes of diabetes. Non-ketotic, antibody-negative mild hyperglycemia in a lean child is one such diagnostic dilemma. Less than 5% of patients with Type 2 diabetes who present at a young age have mild disease, and autosomal dominant transmission. This condition was formerly called maturity-onset diabetes of the young, MODY. It is caused by a single gene mutation and this leads to beta cell dysfunction. Another monogenic defect (not classified under MODY) which impairs beta cell insulin secretion and affects sulfonylurea receptors manifests usually as transient or permanent diabetes with neonatal onset. Other secondary causes of diabetes include chronic pancreatitis and endocrinopathies such as Cushing disease, gigantism and pheochromocytoma. These should also be considered in the differential diagnosis. In this issue, Lal et al., draw our attention to MODY as an important clinical entity through their manuscript on childhood-onset DM. [4] The term ′MODY′ was first coined by Stefan. S. Fajans in 1964, at the Fifth Congress of the International Diabetes Federation in Toronto. [5] Clinically these patients may have subtle manifestations for hyperglycemia and may not require insulin in early years after detection. Till date 11 different types of MODY have been reported and classified as per defects and mutations in beta cell genes. [1] However, due to lack of genetic testing these patients may be over-treated with insulin. The major points of differentiation between Type 1, Type 2 and MODY are: [1],[3],[6],[7]
Genetic testing will disclose whether MODY is present and will help distinguish between subtypes of MODY, and thereby give clues to prognosis and treatment. Once a diagnosis of MODY is made, other family members, both diabetic and non-diabetic, should be screened for family-specific mutation and possible abnormalities of carbohydrate metabolism. Such screening procedures, in ascending order of sensitivity, are as follows: hemoglobin A1C (A1C), fasting plasma glucose, 2-h post-glucose levels, 1-h post-glucose levels, and oral glucose tolerance test (most sensitive). [8] The practical clinical algorithm for evaluation of hyperglycemia in young has been described in [Figure - 1]. Genetics counseling should be provided, especially in children. In case of non-availability of genetic testing trial of sulphonylureas under observation, after ruling out autoimmune pathophysiology constitutes a pragmatic approach. Insulin Promoter Factor-1 (Pancreatic and Duodenal Homeobox 1) subtype MODY-4 Insulin Promoter Factor-1 (IPF-1) gene also known as Pancreatic and Duodenal Homeobox-1 (PDX1) is involved in pancreatic development and beta cell maturation. IPF-1 is homeodomain protein involved in the differentiation of endocrine and exocrine pancreas. PDX-1 acts as major transcriptional regulator of GLUT-2 and GCK (glucokinase) genes in beta cells. IPF-1 homozygous mutations leads to pancreatic aplasia or reduced beta cell number, however, IPF-1 heterozygous mutations lead to reduced beta cell development, reduced glucose sensing to beta cells and reduced glucose metabolism. [1],[7],[9] The current article by Lal et al., has reported their case with IPF-1 mutation in the 5′-UTR associated with hyperglycemia, which also supports non-coding mutations in human diseases. They have clearly characterized and tabulated different forms of MODY in the case report. This mutation was earlier reported by Clocquet et al., in year 2000 in heterozygous members of a family harboring Pro63fsdelC mutation in IPF-1. Their work revealed that the Pro63fsdelC IPF-1 mutation is associated with a severe impairment of beta cell sensitivity to glucose and an apparent increase in peripheral tissue sensitivity to insulin and is a genetically determined cause of beta cell dysfunction. [9] Another pedigree (Michigan Kentucky) with MODY-4 has been described with obesity, hyperinsulinemia and some with Type 2 diabetes (who did not have the mutation in IPF). [10] In PDX1-deficient mice who develop MODY4-type diabetes, an experimental procedure has been discovered that prevents apoptotic and necrotic beta cell death and diabetes. [11] Whether this procedure will be applicable to humans (or other forms of MODY), remains to be determined. References
Copyright 2011 - Journal of Postgraduate Medicine The following images related to this document are available:Photo images[jp11080f1.jpg] |
| |||||||||


{kind=link}