 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Postgraduate Medicine, Vol. 57, No. 4, October-December, 2011, pp. 314-320 Review Article Foothold of NPHS2 mutations in primary nephrotic syndrome AT Jaffer1, WU Ahmed1, DS Raju2, P Jahan1 1 Departments of Genetics and Biotechnology, Osmania University, Hyderabad, Andhra Pradesh, India Date of Submission: 21-Apr-2011 Code Number: jp11088 PMID: 22120861 Abstract Keywords: NPHS2 mutations, podocyte, primary nephrotic syndrome
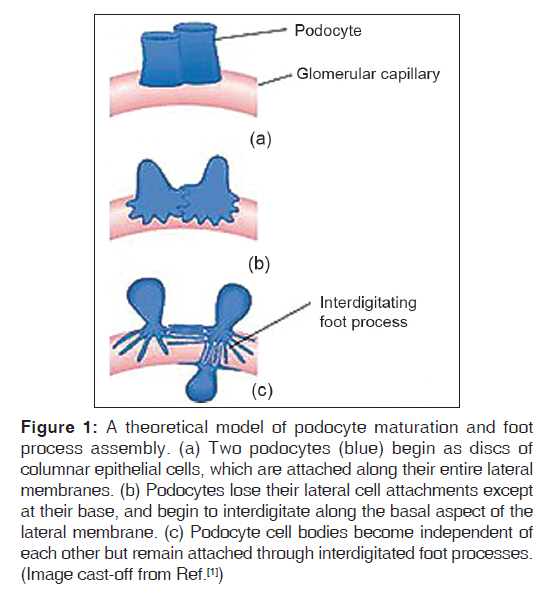
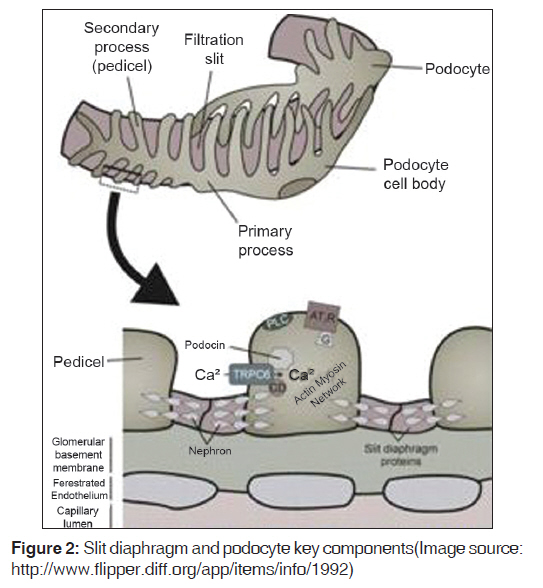
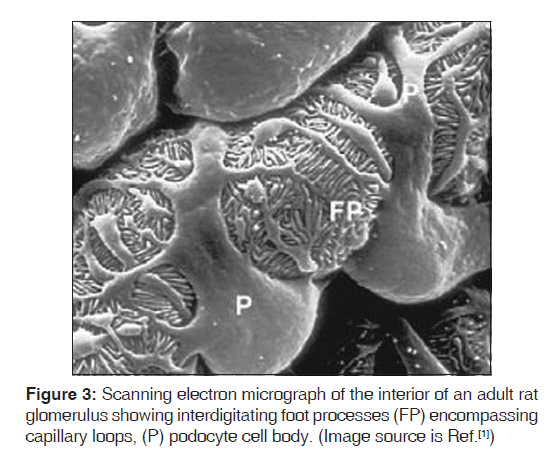
Introduction The glomerulus of the mammalian kidney is a highly developed vascular bed that acts as a filter, allowing a filtrate of small molecules, such as water, sugars, electrolytes, and small proteins, to pass through a barrier that retains high-molecular-weight proteins and cells in the circulation. The proper development and preservation of this structure throughout life is essential for prevention of serious renal disease. Numerous advances in our understanding of glomerular development and function have been witnessed in the past 10 years. Podocytes, the visceral epithelial cell of the glomerulus, are now recognized as being a key cell type, the injury of which can initiate glomerular scarring. Several genetic kidney disorders are caused by mutations in genes that encode proteins that appear to have highly specialized functions in podocytes, especially in the maintenance of the protein barrier, which prevents massive protein loss from the circulation. The glomerular basement membrane and its receptors have also served as one of the key models for the study of how a basal lamina develops and interacts with adjacent epithelial cells. Moreover, glomerular research has added to our understanding of how signals between adjacent cell types are required for the proper development and maintenance of the structural integrity of an organ throughout life. The de novo regeneration of an entire nephron or a whole glomerulus has never been documented in mammals. Indeed, aside from repairing proximal tubules damaged in acute situations, the kidney has a very limited ability to repair itself compared with many other organs. Because the glomerulus only develops in the context of the induction of an entire new nephron during kidney development, it is unlikely that we will learn how to regenerate glomeruli, except in the context of discovering how to regenerate entirely new nephrons. While this remains a long-term goal of kidney development research, perhaps a more accessible therapeutic target will be the podocyte, where an ability to restore foot process architecture has the potential to reduce dramatically the morbidity and mortality that results from chronic kidney disease. [1] We searched Medline and Pubmed using the combination of keywords "NPHS2", "podocin", "steroid-resistant nephrotic syndrome," and "genetics" to identify studies describing an association between NPHS2 gene and chronic kidney disease. Papers published in English between 2000 and June 2009 were reviewed. Reference lists from published articles were also reviewed. We also searched for relevant abstracts and various physiological reviews published by the American Society of Nephrology, International Society of Nephrology, and the International Genetic Epidemiology Society from 2002 to 2008. In this review, we focus on recent advances in glomerular development and biology, and relate them to the disease processes, possible markers, and genetic associations. Cell Biology of the Podocyte Podocytes are highly differentiated cells. They have voluminous cell body, which bulges into the urinary space. The cells give rise to long primary processes that extend towards the capillaries to which they affix by numerous foot processes [Figure - 1]. The foot processes of neighboring podocytes regularly interdigitate, leaving between them meandering filtration slits that are bridged by an extracellular structure, known as the slit diaphragm. The filtration slits are the sites of convective fluid flow through the visceral epithelium. The podocyte is a most spectacular cell type; its location, its architecture, and its relevance are unique. Till date not a single function defined in classic physiological terms can be solidly assigned to the podocyte. It is supposed that (i) it functions as a specific pericyte counteracting the high transmural distending forces permitting the high perfusion of glomerular capillaries and (ii) the podocyte is responsible for continuous cleaning of the filter. [2] Podocytes are polarized epithelial cells with an apical and a basal cell membrane domain. The latter corresponds to the sole plates of the foot processes, which are embedded into the glomerular basement membrane (GBM). The border between the basal and luminal membranes is represented by the slit diaphragm. The luminal membrane and the slit diaphragm are covered by a thick surface coat that is rich in sialoglycoproteins, including podocalyxin, podoendin, and others, which are responsible for the high negative surface charge of the podocytes. [3],[4] Frequently, the apical surface gives rise to a few fingerlike protrusions that float within Bowman′s space; under inflammatory conditions, those processes may be dramatically increased in number and length. The basal cell membrane, i.e. the membrane covering the soles of the foot processes, mediates the affixation to the GBM; it regularly contains coated pits. [5] Both membranes, apical and basal, are heterogeneous with respect to their lipid composition; both contain densely distributed cholesterol-rich domains; [6],[7] corroborating the finding that specific membrane proteins of podocytes are obviously arranged in rafts. [8],[9] The cell body contains a prominent nucleus, a well-developed Golgi system, abundant rough and smooth endoplasmic reticulum, prominent lysosome, and many mitochondria. In contrast to the cell body, indicates a high level of anabolic as well as catabolic activity. A well-developed cytoskeleton accounts for the unique shape of the cells and the maintenance of the processes. In the cell body and the primary processes, microtubules and intermediate filaments, such as vimentin and desmin, dominate, whereas microfilaments, in addition to a thin cortex of actin filaments beneath the cell membrane, [10] are densely accumulated in the foot processes, where they are part of a complex contractile apparatus. Peripherally, the actin bundles appear to be anchored in the dense cytoplasm associated with the cell membrane of the soles of the foot processes. [11] The filtration slits have a constant width of ~30-40 nm [12],[13] and are bridged by the slit diaphragm. The rectangular pores have the approximate size of an albumin molecule. Glomerular filtration barrier and slit membrane Podocytes form a tight network of interdigitating cellular extensions called foot processes, which are bridged by the so-called slit diaphragm. The glomerular filter is composed of the porous endothelium, the GBM, and the podocyte foot processes with the interposed slit diaphragm [Figure - 2]. The filtration barrier is freely permeable by water and small solutes, but to a larger extent, the size selectivity of the filtration barrier for proteins is represented by the slit diaphragms of podocytes. A scanning electron micrograph of the interior of an adult rat glomerulus displays the interdigitating foot processes encompassing capillary loops [Figure - 3]. The slit diaphragm has morphological features reminiscent of adherent junctions, including the presence of a wide intercellular gap and a central dense line. [14] The slit-diaphragm is evocative of a tight junction with differentiated structure and functions. It presents an electron dense zipper-like structure composed of the extra-cellular components nephrin, nephrin homolog-1, Pcadherin, and FAT [15],[16],[17] connected by other specialized structures (i.e., podocin, CD2AP) to the main cell body. [9],[16],[18],[19],[20],[21] Podocin Podocin belongs to the raft-associated stomatin family 41, whose gene NPHS2 is mutated in a subgroup of patients with autosomal-recessive steroid-resistant nephrotic syndrome (NS). Podocin is a membrane-associated protein of the band-7-stomatin family, which interacts with the cytosolic tail of nephrin. Mutations in the NPHS2 gene, localized at chromosome 1q25-q31, cause severe podocyte alterations and NS. [9] It is an integral protein of 383 amino acids with a membrane domain forming a hairpin structure with two cytoplasmic ends at the C- and N-terminus. Oligomerization of podocin clusters and nephrin assembles the slit diaphragm where CD2AP serves as an adapter to the overall network. [22],[23] The presence of an intact podocin is a prerequisite for the transport of nephrin to the membrane and for podocyte intracellular signaling. The NPHS2 coding region is 1,149 bp in length and is followed by a 635-bp 3′UTR containing a typical polyadenylation signals (AATTAAA) situated 13 nucleotide upstream of the poly (A) tail. The gene has eight exons and encodes the 42 kDa integral membrane protein podocin which is expressed in both fetal and mature kidney. While both nephrin and podocin were found to be expressed only in the kidney′s glomeruli, it was also shown that podocin mRNA is expressed in the human fetal heart and was suggested to contribute to normal cardiac development. Primary Nephrotic Syndrome The term "nephrotic syndrome" describes the clinical state characterized by the presence of proteinuria, hypoalbuminemia, and edema. Although other clinicopathologic findings coexist with these three, they remain the central findings in NS. Richard Bright first demonstrated that edema and proteinuria were dependent on changes in the kidney in 1827, and for the next 80 years, NS was known as "Bright′s disease." In 1905, Friedrich von Muller delineated kidney diseases into "nephritis" and "nephrosis", and finally in 1929, Henry Christian included the phrase "nephrotic syndrome" in his writings. [22] In the 18 th and 19 th centuries, treatments for NS included mercury-containing compounds, as well as the induction of malaria or measles. [22] The mortality rate for children with NS in this pre-corticosteroid period was 67%. Then, in 1939 and again in 1944, with the sulfonamides and penicillin, respectively, patients with NS survived longer, and the mortality rate declined to 42% and then 35%, respectively. Finally, in the 1950s, adrenocorticotropic hormone and cortisone became available for the treatment of NS that resulted in a dramatic resolution of proteinuria and the mortality rate decreased to 9%. [23] Idiopathic NS is the most common glomerular disorder of childhood. It is distinct from secondary causes of NS, such as lupus nephritis, hypertensive nephrosclerosis, obesity, IgA nephropathy, diabetic nephropathy, and viral infections, such as hepatitis and HIV. However, the prognosis of NS in children correlates with the spectrum of responsiveness to corticosteroid therapy, from steroid-sensitive nephrotic syndrome (SSNS) to steroid-resistant nephrotic syndrome (SRNS). It is estimated that the annual incidence of NS range from 2 to 7 per 100,000 children, and the prevalence from 12 to 16 per 100,000. This prevalence translates to approx. 1 in 6000 children who have development of NS. Geographic or ethnic differences have been reported with a 6-fold greater incidence in South Asian than in European children. The ratio of males to females with NS is approx. 2:1 in young children, but this male predilection disappears in teenagers and adults. [24],[25] Sex-related differences also exist according to histologic diagnosis. SRNS is the most common acquired cause of end-stage renal disease (ESRD) in children. It accounts for approximately 23% of all pediatric NS and reports indicate that its incidence is still increasing. [26],[27] NS requires the presence of edema, hypoalbuminemia less than 2.5 g/dL, and proteinuria greater than 40 mg/m 2 /h. Remission denotes a reduction of the proteinuria to less than 4 mg/m 2 /h or a urinary albumin dipstick of 0 or trace for three consecutive days. A relapse occurs with the recurrence of proteinuria of greater than or equal to 40 mg/m 2 /h or a urinary albumin dipstick of 2+ or greater. Those patients who go into remission with steroid therapy alone are called steroid responsive, whereas those patients in whom remission is not achieved after 8 weeks of steroid therapy are labeled steroid resistant. [24],[28] One discrepancy in the literature is the lack of a consistent definition of steroid resistance. Patients with SRNS are at risk for extrarenal complications of NS, as well as the progression of their kidney disease to either chronic renal insufficiency (CRI) or ESRD. NS appears to be a clinical heterogeneous condition characterized by histologic variants [26],[27],[29] and different genetic backgrounds [21],[30],[31] with recessive and dominant inheritances. In the last few years, advances in molecular genetics of familial NS have led to the discovery of specialized molecules endowed in podocytes as responsible for proteinuria. They also have clearly indicated that the podocyte is the prevailing glomerular site for repulsion of proteins, a process that allows maintenance of big molecules (such as proteins with a molecular weight >40 kD) in the vascular bed and ultra-filtration of water and solutes. The discovery of mutations of the slit diaphragm components, in particular of podocin, in familial NS represented a break-through in the research of mechanisms of NS that overcome a pure genetic and clinical interest. There is now growing evidence that mutations of slit diaphragm proteins go far behind familial cases and frequently occur in sporadic NS. [21],[32],[33],[34],[35],[36],[37] Pathophysiology Renal biopsy pathologic changes in NS, including minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS), have remained the gold-standard diagnostic classification for decades. Histopathologic changes observed in primary NS renal biopsies are most evident in Light Microscopy (LM) and Electron Microscopy (EM) studies. LM findings in minimal change are generally normal, while in FSGS include focal and segmental glomerular sclerosis, tubular scarring and interstitial fibrosis. EM evaluation of ultrastructural changes often reveals effacement of glomerular epithelial foot processes in minimal change specimens or FSGS. Several histopathologic variants of FSGS have been described, including tip, collapsing, cellular, and classical and perihilar variants. [38],[39],[40] However, none of these variants have been proven to be of prognostic value, particularly in the pediatric population. Numerous animal models of NS exist with histopathological changes consistent with FSGS, including those induced by viral infection or specific medications. Other models demonstrate FSGS associated with altered glomerular hemodynamics secondary to hyperfiltration and nephronopenia, [41] and chronic cyclosporine therapy. [42] While these models provide valuable insight into the pathogenesis of FSGS as a common end-stage glomerular lesion, none are accurate representations of the primary form of SRNS that constitutes up to 15% of cases of ESRD in children. These models are crucial to the study of FSGS that is secondary to known disturbances, such as HIV nephropathy, IV drug abuse, and congenital or acquired nephronopenia. However they are unlikely to help elucidate the etiology of primary NS in children. Structural Biomarkers for Primary NS A routine renal biopsy will yield approximately 8-20 glomeruli, out of a total of 1 million glomeruli normally present in a kidney. If the sample is obtained from an area of kidney that does not contain sclerosis, a patient with FSGS will be diagnosed with MCD. As well, renal biopsies are performed only for children with SRNS and steroid dependence. Children with SSNS are treated with steroid therapy until either (1) the relapses stop occurring, or (2) the disease transforms to frequently relapsing NS or SRNS, in which case a renal biopsy would then be performed. A renal biopsy is therefore an inaccurate test obtained from a skewed population, and cannot provide an appropriate primary NS control group without subjecting SSNS children to a clinically unnecessary and invasive procedure. Although loss of glomerular selectivity for albumin is common to all types of NS, many different and seemingly unrelated genomic and molecular markers correlate with NS disease course, again supporting the molecularly heterogeneic nature of the NS group of disorders. Structural abnormalities in one or more key components of the glomerular basement membrane cause NS, and in some instances, familial NS. Family studies have resulted in the mapping of a gene for autosomal dominant FSGS to human chromosome 19q13 (locus FSGS-1) in a large family from Oklahoma. [43] Genotyping family members at markers on chromosome 19q13 narrowed the candidate interval to a 3.5-Mb region flanked by D19S609 and D19S417. In one additional small family from California, the disease segregated with chromosome 19q13.1 marker alleles. Mutations in the gene encoding a-actinin-4 (ACTN4) on chromosome 19q13.1, an actin filament crosslinking protein, were discovered as the cause of FSGS in families with an autosomal dominant form of FSGS. [44] Based on actin-binding experiments, it was shown that these mutations result in altered regulation of the actin cytoskeleton of glomerular podocytes, the epithelial cells that play a key role in the glomerular basement membrane. A second locus for dominant FSGS has been identified on chromosome 11q22-24 in one family [45] and a recessive locus has been reported on chromosome 1q. [46] Congenital NS of the Finnish type has been shown to be the result of defects in both alleles of the NPHS1 gene, which encodes nephrin, a transmembrane protein located at the slit diaphragm. [37] Recently, a mutation in the NPHS2 gene encoding the glomerular epithelial cell component, podocin, is the cause of FSGS in a subgroup of affected families. NPHS2 mutations in 30 families with autosomal recessive SRNS revealed that 9 of the 30 families were found to have NPHS2 mutations that were the likely cause of recessively inherited NS. For six of these nine families, affected individuals were compound heterozygotes for a non-conservative R229Q amino acid substitution in the podocin protein. [47] NPHS2 Mutation in Familial and Sporadic NS Podocin is encoded by the gene NPHS2 that was discovered and localized at 1q25 by positional cloning in 2000. [25] Mutations of NPHS2 have been identified in several families with autosomal recessive-FSGS (ARFSGS). This accounts for most of familial NS with recessive inheritance, and in sporadic cases as well. Overall, results from the studies above demonstrate that NPHS2 mutations account for a significant part of all nephrotic patients roughly corresponding to a mutation detection rate of 45-55% in families with recessive traits and 8-20% in sporadic NS. However, the incidence rate would increase if we consider only patients with poor response to steroids and/or showing pathology of FSGS. It must be clearly stated that all cases with sporadic NS and NPHS2 mutation are, in fact, familial and this high incidence underscores a comparably high frequency of healthy carriers at least in the geographic areas where screenings were done. More than 50 NPHS2 mutations have been reported that occurred as homozygous or compound heterozygous combinations in 79 patients with a familial trait and 54 sporadic cases. Reported mutations involve the whole length of the gene and determine every kind of alteration, including missense, nonsense, and deletion. Studies so far published refer to European populations and report different distributions of mutations: R138Q was most frequently found in Germany and France [34],[35] while the P20L variant was observed mainly in Italy [32] and Turkey. One unique case involves the presence of heterozygous NPHS2 mutation associated with the R229Q variant that is a polymorphism with functional effects. This association was found in 33 familial cases and in 2 sporadic NS. Age at onset of proteinuria was rather variable in different reports, in general occurring before the 10 th year with a strong prevalence of pediatric patients. A few cases with congenital or very early NS were described in all patient groups, including two families with double homozygous R168H and P20L mutations. It has been reported that the presence of two recessive NPHS2 mutations caused SRNS in 18.1% (73 of 404) families and that there is statistical significance for early onset of SRNS in children with truncating or homozygous R138Q mutations of NPHS2 (<1.77 year) versus children with all other sequence variants (>4.17 year). This finding will be important for the prognostic evaluation and management of renal replacement therapy in children with SRNS. The vast majority (56 [98.2%] of 57) of children with two truncating or homozygous R138Q mutations presented at <6 year of age. Truncating or homozygous R138Q mutations are frequent pathogenic NPHS2 mutations. [48] A study by Hildebrandt et al. and several other authors suggest that children with a first episode of NS should be tested for podocin mutations before therapy to avoid an unnecessary steroid course in those with NPHS2 mutations. Moreover, there is a low rate of recurrence in patients with podocin mutations, suggesting that circulating factors are not involved in the pathogenesis of NS in patients with a molecular defect of a podocyte protein.
The glomerulus of the mammalian kidney is an intricate structure that contains an unusual filtration barrier that retains higher molecular weight proteins and blood cells in the circulation. Recent studies have changed our conception of the glomerulus from a relatively static structure to a dynamic one, whose integrity depends on the interactions between key components of the visceral epithelial cells-podocytes. Research into the mechanisms that control glomerular development and then maintain glomerular integrity and function has recently identified several genes that are mutated in human kidney disease. The discovery of molecular defects of podocyte components causing familial forms of NS suggest the role of podocytes as the site of permselectivity in the kidney. Since then, knockout models of podocyte components and further molecular genetic screenings definitely consolidated this concept. Podocin is an integral membrane protein of podocytes that interacts with nephrin and contributes to maintain the intact ultra-filtration barrier at the slit-diaphragm. The discovery of mutations of NPHS2 in familial NS represented a breakthrough in the research of mechanisms of proteinuria that overcomes a pure genetic and clinical interest. NPHS2 mutations account for a significant proportion of all nephrotic patients, roughly corresponding to a mutation detection rate of 45-55% in families with recessive traits and 8-20% of sporadic cases, according to different cohorts and considering all the clinical phenotypes. Podocin was the second recognized protein to cause proteinuria in familial cases with recessive inheritance and in sporadic patients. The clinical picture of NS caused by podocin mutations ranges from an early onset, thus resembling congenital NS of Finnish type, to a late onset in the second decade of life, resembling idiopathic FSGS. Several NPHS2 mutations have been reported in both familial and non-familial SRNS, which involve the whole length of the gene. Published studies report different distributions of mutations, and these variations in frequency and type of NPHS2 mutations among different populations may partially explain the inter-ethnic difference in the prevalence as well as the outcome of SRNS. Thus, podocin is turning out to be a major contributor to the genetic burden of NS. Only a molecular approach is diagnostic in this setting, and it is proposed that all cases with heavy proteinuria that is resistant to steroids and suggestive of FSGS should undergo genotyping of NPHS2 before planning therapeutic regimens with steroids and other immunosuppressive drugs. [49] To our knowledge, however, whether or not NPHS2 is the causative gene in Indian SRNS has not been established (work in progress by our group). Several familial cases have been observed in pediatric renal clinics in several distinct geographical regions of India that may give a clearer picture of the genetic epidemiology of this problem in our country. Recognition of the genetic origin of disease and detection of the resultant mutation(s) shall be useful clinically. This might allow clinicians to avoid unnecessary and aggressive immunosuppressive treatments, to predict the absence of recurrence after transplantation, and to provide prenatal diagnosis to families at risk. References
Copyright 2011 - Journal of Postgraduate Medicine The following images related to this document are available:Photo images[jp11088f1.jpg] [jp11088f3.jpg] [jp11088f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}