 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
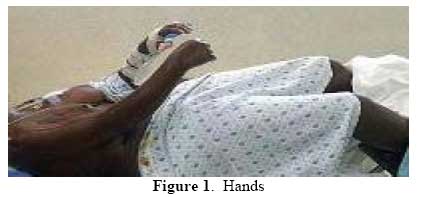
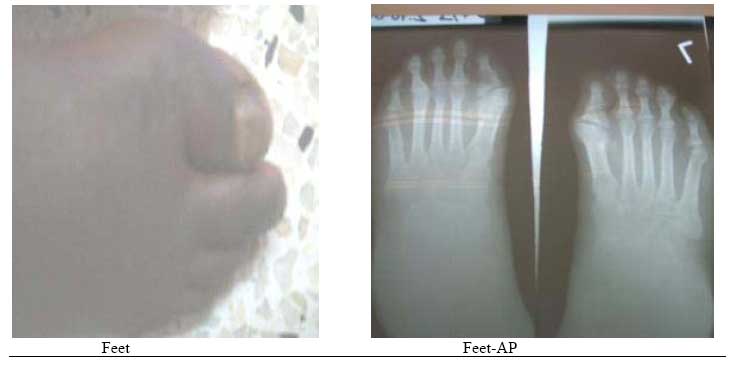
East and Central African Journal of Surgery, Vol. 12, No. 2, November/December 2006, pp. 95-98 Fibrodyslasia (Myositis) Ossificans Progressiva- A Case Report E.N. Muteti, T.C. Mead. Code Number: js07048 We are presenting a case report of an 18yr old kenyan girl with big toe/ thumb anomalies and a 3 year history of heterotopic ossification of the temporo-mandibular joints, cervical spine, shoulders, elbows, lumbar spine and abdominal wall. This case highlights the clinical features of this rare disorder as seen in this patient, onset at 14years of age and presentation.Case Presentation History An 18yr-old girl presented with a year history of a swelling around the temporo-mandibular region that was followed by difficult in opening her mouth. In the following 12 months after the onset, she developed painless and progressive stiffness of the neck, then the shoulders, elbow and hips in that order. She could not fully open her mouth, was unable to use her upper limbs for activities of daily living or walk upright. She gave no history of fever, pain, weight loss or trauma. The respiratory, cardiovascular, gastrointestinal, genitourinary and nervous systems were normal. There was no history of a similar disease in the family and she was the product of a nonconsanguinous marriage. Findings on ExaminationExamination revealed a well-nourished girl, not pale, normal skin,normal vital signs and walks with a crouched gait. The mouth opening distance was 2cm. She had limited range of motion of the neck of 10 degrees flexion, no rotation or lateral flexion. The shoulder were held in fixed 10 degrees of abduction and 30 degrees of flexion. The elbows were held in fixed flexion at 20 degrees on the right and 45 degrees on the left. The right forearm was in fixed 20 degrees of pronation, while the left was held in full pronation. The hips had flexion deformity of 20 degrees but normal rotation. The thumbs and big toes were abnormally short. The range of motion of the wrists, fingers, knees, ankles and toes were normal. The right half of the patients abdominal wall felt rock-hard on palpation while the left was normal. InvestigationsThese included:
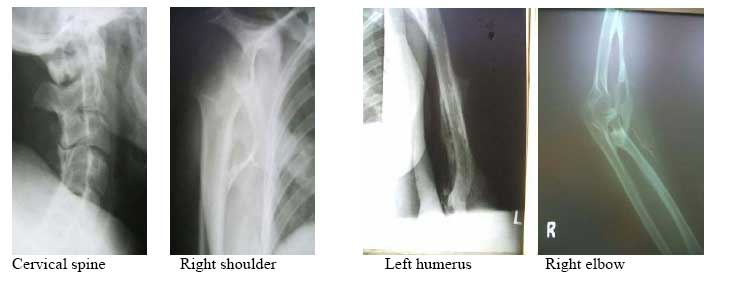
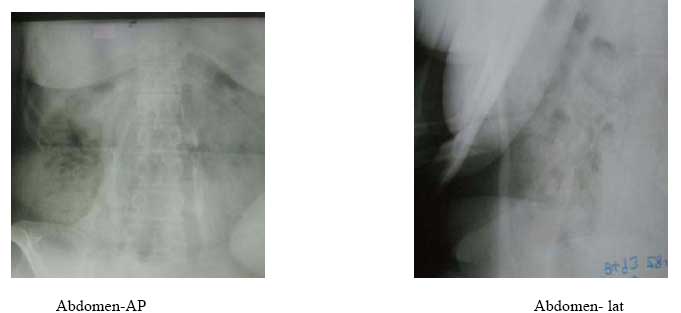
X-ray findings: bone bridge between scapula and humerus (shoulder), humerus and ulna (elbow) and along the triceps and biceps, bony ankylosis of C2/C3, C4/C5 posterior spinous processes; and bone formation along the lumbar paraspinal muscles and anterior abdominal wall. DiscussionFibrodysplasia ossificans progressiva, also known as Myositis ossificans progressiva, is an extremely rare disease. The reported prevalence rate in Britain and the United States of America is 1 in 2 million people. It is estimated that there are only 2 500 people suffering from this disease worldwide1 . 600 of these are known to the investigators of this disease at the University of Pennsylvania2 . Kaplan ,Kitterman and Kantanie3 studied 138 members of the International Fibrodysplasia Ossificans Progressiva Association; and found out that the initial diagnosis was incorrect in 87% of cases. The mean period of onset to correct diagnosis was 4.1 years and a median number of 6 physicians had been consulted up to the point of diagnosis. Unnecessary biopsies and therapy was given in 67% and 68% of patients respectively. There was reported loss of mobility after invasive intervention in 49% of cases studied. We consequently present this case found in our region to highlight the unique features of this rare disease. There was a delay in diagnosis of 3 years in this patient and surgery (biopsy) had been done on the left elbow prior to been seen in our hospital. Connor and Evans4 reported the clinical features and natural history of this disease in 34 patients. He noted that the disease usually starts in childhood by 10 years of age(up to 25years of age) with swelling and later stiffness due to heterotopic bone formation in skeletal muscle. This began at the neck, progressed caniocaudally (to involve the back, hips and knees) and centripetally (to involve shoulders, elbow and wrist). The patients had congenital malformation Abdomen- lat of the big toe and thumb (microdactyly, hallux valgus, symphalangism, delta phalanx) which is key to the diagnosis. In our patient the onset was at 15 years of age with temporo-mandibular joint as the initial site of involvement. This is in keeping with Marranes et al observation, that jaw fixation is key to the diagnosis of fibrodysplasia ossificans progressiva5. She had the classic cranio-caudal and centripetal sequence of joint involvement, thumb and big-toe abnormalities, and extensive heterotopic bone in the muscle without intra-articular involvement. The swelling/stiffness involved multiple joints, was painless, not associated with fever or trauma; thus excluding myositis ossificans circumscripta. Inaddition to the classic big toe anomaly, there was no skin involvement of the skin in our patient,thus excluding progressive osseous heteroplasia and osteoma cutis. Albright's renal osteodystrophy was unlikely in the absence of renal disease symptomatology. The laboratory investigations were normal as expected6. In early disease the alkaline phosphatase level is elevated. In late disease, there may be conduction block in the heart. The radiographs showed extensive bone formation in the skeletal muscles of the upper limbs, abdominal wall and spine. Shore, Feldmann, Kaplan et al7 have shown that the cause of fibrodysplasia is a mutation in the bone morphogenetic protein type 1 receptor, ACVR1, in chromosme 2. This is a single nucleotide substitution in the ACVR1 gene of arginine instead of histidine in position 206. The mutation causes abnormalities in the BMP signalling pathway and heterotopic ossification in skeletal muscle. Our patient has multiple problems arising from the immobilization of multiple joints by the heterotopic bone, including difficult in opening the mouth, upper extremity use and walking. The treatment of this disease is difficult.Various modalities have been used including surgery, anti-inflammatory medications (NSAIDs principally indomethacin; corticosteroids), biphosphonates, exercises, irradiation, and bone marrow transplantation. The results have been disappointing. Current reports of success in literature suggest combination therapy using indomethacin, biphosphonates and single dose radiotherapy8,9,10,11. ConclusionFibrodysplasia ossificans progressiva is a very rare disease. We present the clinical features of one such case found in our region. The unique features are: initial site of involvement is the temporomandibular joint, painless swelling/stiffness of involved areas and onset of disease at 15years of age. This disease is characterized by cranio-caudal and centripetal sequence of heterotopic ossification and big toe abnormalities. [ Fig.1 ] [ Fig. 2 ] [ Fig.3 ] [ Fig.4 ] [ Fig.5 ] References
© 2007 East and Central African Journal of Surgery The following images related to this document are available:Photo images[js07048f2.jpg] [js07048f3.jpg] [js07048f1.jpg] [js07048f5.jpg] [js07048f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}