 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
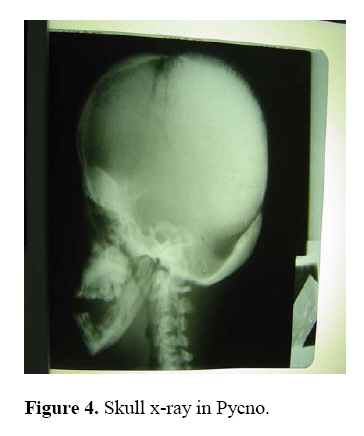
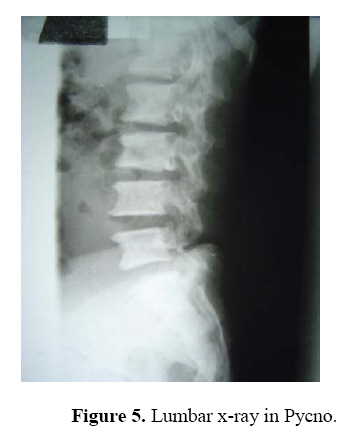
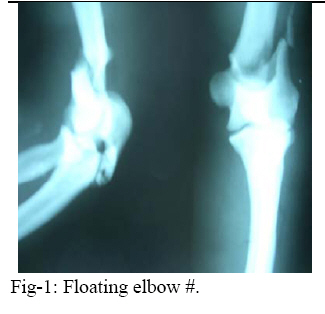
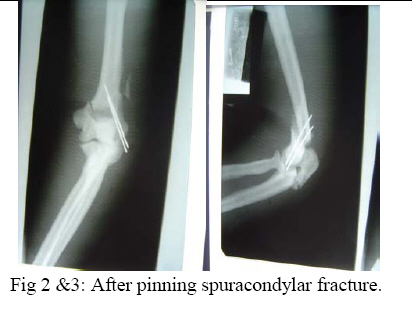
East and Central African Journal of Surgery, Vol. 14, No. 1, March-April 2009, pp. 98-102 Pycnodysostosis with Epilepsy in a Malawian patient: A Case Report B. L. Wamisho1, J. Bates2 1Addis Ababa University, Department of Orthopedics, Addis Ababa, ETHIOPIA, 2College of Medicine, Blantyre, Malawi, Code Number: js09017 Background: Pycnodysostosis is a very rare genetic disease resulting from deletion the short arm of one of the group – G chromosomes leading to deficiency of Cathepsin –K, a cysteine protease needed for proper functioning of Osteoclasts to resorb and remodel bone. Unlike many skeletal dysplasias, it is lysosomal enzyme defect and it is inherited in a typical autosomal recessive mode. Patients present with repeated fractures due to brittle bone as well as multiple other health problems. World wide extremely few cases have been reported. This presentation therefore is aimed at documenting an occurrence of a very rare disease from Blantyre, Malawi. We hope that this report creates awareness among clinicians in dealing with some 200 various types of skeletal dysplasias, and the diagnosis should be considered when evaluating patients with strikingly short stature. This is a typical case description of the syndrome, including clinical approach and radiological features of a patient seen at College of Medicine, Queen Elisabeth Central Hospital (QECH), in January 2009. Introduction The Greek word ‘Pycno’ in Pycnodysostosis means dense, thick or compact. Pycnodysostosis consistently causes short stature. The height of adult males with the disease is less than 150 cm (59 inches, or 4 feet 1 inch). Adult females with Pycnodysostosis are even shorter. In Pycnodysostosis the bones are abnormally dense (osteosclerosis), the distal phalanges of both fingers and toes are unusually short, and there is delayed closure of the skull sutures in infancy so that the fontanels remain widely open. The sclerotic bones are brittle and fracture easily. Pathological fractures occur more commonly in the long bones of the lower limbs and feet, as well as the mandible and clavicle. Pycnodysostosis can be grouped with other genetic diseases that are individually uncommon, but collectively important because of the sum of their numbers, their heavy impact upon affected individuals, and the equally heavy burden they place upon their families. Pycnodysostosis is a very rare genetic disease resulting from deletion the short arm of one of the group – G chromosomes (21 and 22 and the Y chromosome pairs), inherited in a typical autosomal recessive mode. In 1995, the gene was first charted by Gelb and associates. It was found to travel preferentially with gene markers known to be in chromosome region 1q21. In 1996, patients with Pycnodysostosis were shown by Gelb and coworkers consistently to have mutational changes in the gene for Cathepsin K. The defective Cathepsin K gene was thus demonstrated to be the gene responsible for Pycnodysostosis. Pycnodysostosis is now clearly recognized as being due to Cathepsin K deficiency. Cathepsin K is an enzyme (a catalyst for a reaction of body metabolism) of the type called a cysteine protease. This protease is important in the cells of normal bone (Osteoclasts) that are responsible for bone resorption. It is thought that the normal function of Osteoclasts is impaired in patients with Pycnodysostosis due to a lack of Cathepsin K and there is a failure to adequately reabsorb that component of bone called the organic matrix. (This process is essential for maintenance of normal bone; a process referred to as remodeling). Because of this inadequate resorption, the bones in Pycnodysostosis are abnormally dense and brittle. Due to repeated fractures or other health problems, patient with Pycno are likely to present on a regular basis to health institutions. The precise frequency of Pycnodysostosis has never been determined and the disease is internationally classified as a ‘2 star rare disease’. The concomitant occurrence of epilepsy in our Pycnodysostosis patient from Malawi is rare and makes this an important case that should be reported to the international medical community. Case Report A 38 years old Malawian male with dark African complexion presented to QECH with a left combined supracondylar and olecranon fracture which he had sustained after a simple twisting injury. It is the 7th fracture in the last ten years. The patient has previously sustained fractures in both femurs and tibias, one tibia twice. He stopped his job as a ‘dancer’ due to repeated fractures from simple twists or falls. He has also sustained a clavicle fracture and now this elbow injury. It happened as a result of a simple twisting injury that occurred during a disagreement with a taxi driver. In situ pinning of the fracture was successfully done. The patient is also a known to have epilepsy and has been on treatment since childhood. He had repeated tooth extraction and undergone major dental surgery. He is the shortest of all the 7 siblings in a family of 9 and is the only one affected by the condition. Parents are normal and there were no stillbirths or abortions. Family members reported that he was slower in achieving developmental milestones. He denied bleeding tendency, repeated infections or symptoms of anemia. He said he was poor at school performance. He is a married merchant with no sexual dysfunction. Physical examination revealed a man with strikingly short stature (1.29 meters) and proportionate dwarfism. He has characteristic frontal bossing with narrow mid face, and both fontanels are open. He has white sclera, pink conjunctiva and no hearing problems. All cranial nerves are intact. He is slow in speech & looks to have moderate retardation.(not sure I would agree that he is retarded) His chin is very small and he has nasal speech, small, irregular decayed teeth with evidence of multiple extractions.. The palate is high arched and looks grooved (anesthetic intubation was difficult). The clavicles are underdeveloped. Chest, abdominal & genitourinary examinations were normal and the spine is straight. The hands are very short especially the distal phalanges, the overlying skin is wrinkled. Nails are just visible. All limbs are extremely short deformity consistent with previous fractures and there is no significant bowing. All joints are normal. Finally all available family photographs were inspected and he is the only one with strikingly short stature. Radiological skeletal survey revealed: generalized increased bone density in all bones, Signs of multiple healed fractures in the long bones, widely open fontanels and sutures with very small maxilla, poor dentition (Figure 4). Clavicles are hypoplastic with acromioclavicular joint laxity. The chest x-ray is normal. The lumbar vertebrae are short with wide intervertebral disc spacing (Figure 5). There is a combined supracondylar and olecranon fracture of the left elbow. The supracondylar fracture was wired in situ and the olecranon fracture was left untouched for there is good callus & union observed intra-operatively. All x-rays were discussed with radiologist. (Figure 1, 2-3) Hematological investigations revealed normal white cell, red cell & platelet counts. His hemoglobin is 11.4 mg%. Serum calcium is normal & alkaline phosphatase level is also with in normal range. Serial follow-ups revealed that the fracture has united at the normally expected time. The elbow range of motion is reasonably good. Two similar cases have been seen in the department over the last 10 years but none was epileptic. Discussion Causes of strikingly short stature are too many (over 200 forms of skeletal dysplasias) and diagnosis of the specific syndrome, with out the help of a molecular genetic labs is at times difficult2. Yet, combined clinical knowledge, systematic (algorism) approach, simple blood tests and imaging may lead to reasonable differential diagnosis3,4,5,6. Of all causes of dwarfism, the only two diseases that are associated with a generalized increase in density of bone are osteopetrosis and Pycnodysostosis. Both result from impaired function of Osteoclasts and lead to “brittle” dense bones due to defective resorption of matrix8 . Patients with osteopetrosis display macrocephaly, progressive deafness and blindness, hepatoplenomegaly, tetany, and severe anemia beginning in early infancy or in fetal life. Deafness and blindness are generally thought to represent effects of pressure on nerves. The anemia is caused by encroachment of bone on marrow, resulting in obliteration, and the hepatoplenomegaly is caused by compensatory extramedullary hematopoiesis. Anemia is a rule in long-standing osteopetrosis but very rarely presents with Pycnodysostosis1,7 . With any of these findings lacking in our patient and working out through dysplasia algorisms in textbooks, we can confidently diagnose him as a case of Pycnodysostosis-clinically. Biopsy is not essential. Pycnodysostosis (also called Toulouse-Lautrec syndrome) is perhaps best known as the diagnosis given retrospectively to the late 19th century French artist Henri de Toulouse-Lautrec (portrayed by Jose Ferrer in the 1952 film "Moulin Rouge")9. It is a very rare lysosomal enzyme defect disease due to mutation of Cathepsin-K gene on short arm of group Gchromosomes (21, 22 & Y) leading to failure of Osteoclasts to model & re-model bone. The unresorbed mineral matrix makes the bone dense but brittle as chalk –causing recurrent fractures easily; hence such patients should avoid combative sports10 . Pycnodysostosis with epilepsy is rare and has been reported as a syndrome known as Gurrieri syndrome. This is characterized by a combination of epilepsy, short stature and skeletal abnormalities11,12,13. The cause is not clearly known but thought to be due to calcifications in the brain or malformed/shaped skull. Our patient has been seen in the epilepsy clinic at QECH since childhood. He takes regular phenobarbitone and phenytoin occasionally. He discontinued drugs briefly & has suffered from a generalized tonic-clonic seizure. The challenges in operative fracture treatment of these patients are twofold. There may be difficulties with intubation due to the small mandible, dental problems and high arched grooved palate. There maybe difficulty in drilling the bone or inserting K wires due to the dense bone. Otherwise, the healing of the fractures is uneventful and will proceed like a normal bone. Conclusion This is probably the first African report of an extremely rare case of Pycnodysostosis with Epilepsy involving an adult male black African. The report reminds us once again of the existence of this rather rare disease entity Africans. The general objective of this report is to create awareness of the existence of other causes of strikingly short stature other than Achondroplasia, especially when they display recessive inheritance. It also encourages the clinician to systematically approach a complex group of diseases and reach to a reasonable differential diagnosis based on limited, simple, basic investigations14 . References
© 2009 East and Central African Journal of Surgery The following images related to this document are available:Photo images[js09017f5.jpg] [js09017f1.jpg] [js09017f2-3.jpg] [js09017f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}