 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
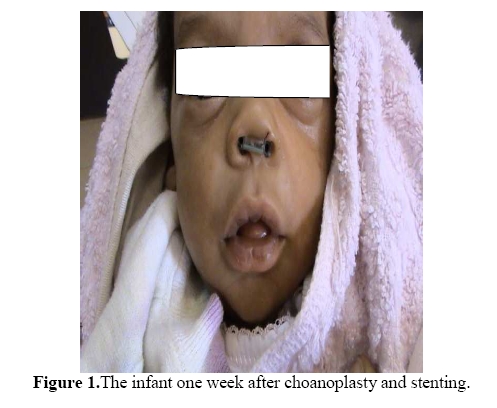
East and Central African Journal of Surgery, Vol. 15, No. 1, Mar-Apr, 2010, pp. 93-95 Case Report Choanal Atresia in Siblings: Case reportKaitesi B.M. Otolaryngology Department, University Teaching Hospital, Kigali – Rwanda. E-mail: kaibat@hotmail.com Code Number: js10015 Choanal atresia is an uncommon and often poorly recognized cause of unilateral or bilateral nasal obstruction. This report describes the case of bilateral choanal atresia in two consecutive siblings and describes the methods of treatment offered. Case Report A female infant born at term by spontaneous vaginal delivery was brought to the Otolaryngology Department two hours after birth with difficulty in breathing. The infant was born by a 24- years old woman, gravida 2 para 1. On arrival in the Otolaryngology Department, the infant was noted to have generalised cyanosis. Detailed physical examination revealed an infant at term, normal for gestational age but with severe respiratory distress. The vital signs were normal except for oxygen saturation which was below 85%. Tachypnoea was also noted. Marked intercostal and subcostal retractions were noted but vesicular breath sounds were perceived and were symmetrical. A 3.5mm suction tube could not be passed through either nostril. On placement of an oral-pharyngeal airway breathing improved significantly and the cyanosis decreased till it disappeared. Urgent choanoplasty and placement of stents was done and the patient had an uneventful recovery (Figure 1). The infant was followed up weekly then fortnightly then monthly. At 3 months, the stents were removed and the patient has fared on very well. During history taking, it was established that the same woman had been admitted eleven months before with her first born. We reviewed our records and found out that a term infant was brought to our department at the similar timing and presented with similar symptoms like the second child immediately after birth. This was a female infant on whom choanoplasty was done for bilateral choanal atresia. Stents had been removed six weeks after placement and the child was doing well. During her last visit, both children were examined. No gross congenital anomalies were found except a single pre-auricular pit found on the right side in both children. Discussion Choanal atresia was first described by Roederer in 1775, and was first reported in Britain in 1881 by Ronaldson. Carl Emmert, in Bern, operated successfully on a patient with choanal atresia in 1851.1 Choanal atresia is a rare condition with an incidence of 1 in 7000 live births. It is believed to be as a result of persistence of the buccopharyngeal membrane during the embryological period. The genetics remain unclear.2In one review, the family histories of patients with choanal atresia revealed no obvious hereditary trend, and a chromosome analysis showed no abnormalities.3 This results in complete obstruction of the posterior nasal openings in one or both sides. This condition usually occurs sporadically, but has been described in siblings and successive generations.2 The blockage may be either bony or membranous. A mixed picture is usually seen in up to 70% of cases. It affects women more frequently than men in the ratio 2: 1; there are 3 unilateral cases of choanal atresia for every two bilateral. Bony atresia is far more common than membranous atresia, accounting for 90% of reported cases 4. Bilateral choanal atresia will present as an acute emergency since neonates are obligate nasal breathers. The classical picture of cyclical cyanosis which is relieved by crying is usually seen.1 Placement of a metallic spatula just below the child’s external nasal aperture helps to exclude choanal atresia if misting occurs unilaterally or bilaterally. Failure to pass a nasal catheter suggests atresia which can be confirmed on nasal endoscopy or CT Scanning. Choanal atresia may be isolated or a feature of associated congenital anomalies. However 60% of cases of choanal atresia have an associated congenital defect. It has been found to be associated with syndromes such as Downs and Treacher-Collins. It may however, be found with other isolated defects such as palatal cleft, high arched palate, micrognathia, tracheoesophageal fistula, missing teeth or facial cleft. Choanal atresia has been linked with a collection of defects - CHARGE association.5, 6 These anomalies should be excluded in subjects with choanal atresia. One should search for other congenital defects of the heart, eyes, and gastrointestinal and urinary tract whenever a diagnosis of choanal atresia is documented. An ophthalmologic and audiology review is also necessary. Unilateral choanal atresia may present late in life with symptoms of persistent unilateral nasal discharge. A foreign body should be excluded. Management is purely surgical by a transpalatal or transnasal by endoscopy approach.7 The outcome and superiority of the surgical approaches is still under scrutiny since no comparative studies have been conducted yet. Differential diagnoses include deviated nasal septum, dislocated nasal septum, septal hematoma, mucosal swelling, encephalocele, nasal dermoid, hamartoma, chordoma, hypertrophied turbinate and teratoma. Conclusion In acute respiratory distress in a neonate, bilateral choanal atresia should be considered. It is a medical emergency. The diagnosis is easily made with a small catheter; a tongue blade and feeding tube should be used for diagnosis. Anatomical confirmation by radiographs should be made. Whenever one congenital anomaly is found, others should be sought. Surgical correction is required. Heredity of choanal atresia has been disapproved in most studies conducted on animals and this has been projected in humans. In our case report, consecutive siblings had bilateral choanal atresia. Is this hereditary or an incidental finding? References
Copyright © 2010 - East and Central African Journal of Surgery The following images related to this document are available:Photo images[js10015f1.jpg] |
| |||||||||

{kind=link}