 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Archivos Latinoamericanos de Produccion Animal, Vol. 13, No. 1, Enero-Abril, 2005, pp. 30-42 Los microsatélites (STR's), marcadores moleculares de ADN porexcelencia para programas de conservación: una revisión Microsatellites (STR's), ADN Molecular Markers for Excellencyfor conservation programs: A review J. A. Aranguren-Méndez1, R. Román-Bravo1, W. Isea1, Y. Villasmil1, y J. Jordana2 Facultad de Ciencias Veterinarias, La Universidad del Zulia. Apartado 15252, Maracaibo, 1 Enviar correspondencia a E-mail:
atilioaranguren@icnet.com.ve
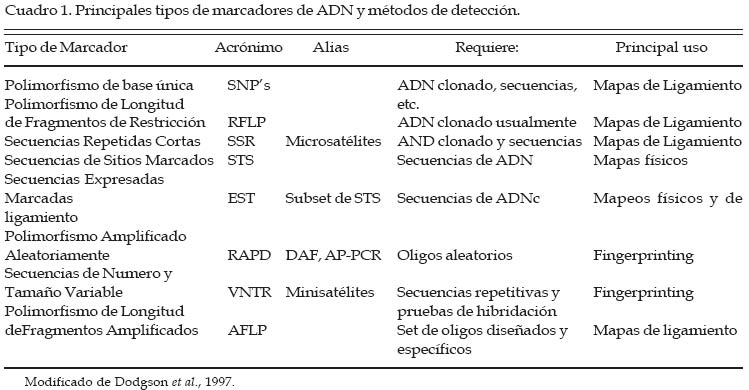
Recibido Octubre 21, 2003. Code Number: la05005 ABSTRACT Keywords: Microsatellite, DNA, polymorphism, conservation RESUMEN Palabras clave: Microsatélite, ADN, polimorfismo, conservación. INTRODUCCIÓN En genética, tanto desde el punto de vista de la las nuevas herramientas estadísticas han permitido mejora como de la conservación, el principal objetivo la posibilidad de identificar y utilizar esa variación que se persigue es estudiar, determinar y medir la genómica para la mejora del ganado. Desde el punto variación existente, entre y dentro de los individuos de vista molecular, esta variación, también denominada «polimorfismo» derivada de cambios espontáneos en el ADN puede medirse hoy día a través de diferentes técnicas disponibles. Los cambios observados van desde la substitución de un simple nucleótido, que representa el tipo más común de mutación y que puede ser detectado a través del estudio y análisis de los polimorfismos de base única (SNP), hasta mutaciones que involucren mayores números de sitios nucleotídicos (Dodgson et al., 1997). Para efectuar estos estudios, ha sido de gran importancia el desarrollo de la técnica de la reacción en cadena de polimerasa (PCR) por Mullis et al. (1986), la cuál es una reacción enzimática catalizada por una ADN polimerasa, que permite la amplificación o reproducción «in vitro» de grandes cantidades de una región particular del ADN, a partir de una pequeña cantidad original de ADN molde. Esta enzima requiere, para la síntesis, de un par de oligos denominados iniciadores o «primers», cuyas secuencias son complementarias a la de las regiones flanqueantes 5' y 3' del segmento particular de ADN que se aspira amplificar. Los marcadores de ADN, y en especial los microsatélites, están rápidamente reemplazando o complementando a otros marcadores o metodologías genéticas en las aplicaciones evolutivas y la conservación de las especies. Dado su elevado nivel de polimorfismo, resultan útiles para definir un único genotipo multilocus, de particular interés en estudios donde se requiera una escala muy fina de resolución y en los cuáles otros tipos de marcadores podrían presentar algunas limitaciones, siendo especialmente estos estudios los referidos a: análisis de paternidades, parentescos, asignación de individuos a raza, planificación de apareamientos y otros aspectos de índole reproductiva (Jones et al., 1986). Un buen marcador molecular debe reunir una serie de características para maximizar su utilidad, entre ellas, debe tener una buena distribución a lo largo del genoma y alto grado de polimorfismo. La técnica para analizar el marcador debe ser rápida, práctica y debe repetirse con fiabilidad en otros laboratorios (Cheng y Crittenden, 1994). Variación genética y métodos de detección Desde la aparición de la PCR se han desarrollado una serie de técnicas utilizadas para los estudios de variabilidad genética en las especies animales. Entre ellas técnicas se cita el uso de los marcadores genéticos (Cuadro 1), la cual se basa en la clonación y secuenciación de fragmentos de ADN, mientras que otros, más aleatorios, se basan en la detección de polimorfismos al azar (fingerprint markers o huellas digitales moleculares) (Dodgson et al., 1997). Algunas características y particularidades notables de los principales marcadores moleculares más utilizados en genética de poblaciones se detallan a continuación: SNP (polimorfismo de base única) Es un tipo de polimorfismo que corresponde a la diferencia en una simple posición nucelotídica, por ejemplo sustituciones, delecciones o inserciones de un solo nucleótido. La mayoría de este tipo de polimorfismo presenta sólo dos tipos de alelos (bialelica), y por ello son referidas algunas veces como marcadores bialélicos. Con la presencia solamente de dos alelos, la máxima heterocigosidad esperada para cada SNP es de solamente 50%, siendo por lo tanto menos informativo que las regiones satélites de ADN (minisatélites y microsatélites), las cuales generalmente presentan múltiples alelos y sus valores de heterocigosis superan el 70%. En forma general, para la construcción de mapas genéticos a partir de SNP son requeridos hasta 3 veces mas marcadores comparativamente que con los STRP's (Kwok et al., 1996). Aunque muchos de los SNP's se localizan en regiones no codificantes, un número importante de estas mutaciones que corresponden a sitios de genes (codificantes) se han asociado a enfermedades u otra expresión fenotípica. Su alta frecuencia en el genoma y su baja tasa de mutación, hacen que se constituyan como elementos deseables para la construcción de mapas genéticos. En el caso del genoma humano, donde mayoritariamente se han estudiado los SNP's, se estima que un SNP con una heterocigosis por encima del 30% se espera que se presente cada 1.3 kb (Kwok et al., 1996). Minisatélites o Número de Secuencias de Tamaño Variables (VNTR) Marcadores polimórficos descubiertos por Jeffreys et al. (1985), en el que ciertas pruebas de hibridación para secuencias repetitivas generaban un complejo patrón de bandas que contenían un polimorfismo heredable. Los VNTR son repeticiones al azar en tándem de 10 a 60 pb, altamente polimórficos y con elevadas tasas de heterocigosis en las poblaciones (Vance y Othmane, 1998).Microsatélites o Secuencias Repetidas Cortas (SSR) Los microsatélites son segmentos cortos de ADN de 1 a 6 pares de bases (pb), que se repiten en tándem y de forma aleatoria en el genoma de los seres vivos (Figura 2). Algunas de las características de estos marcadores versus otros (minisatélites, RFLPs, RAPDs, etc.) que son considerados por la mayoría de autores como una poderosa herramienta para estudios genéticos son: presentan un elevado grado de polimorfismo, su herencia es mendeliana simple, son codominates (pudiéndose diferenciar los individuos homocigotos de los heterocigotos), son fáciles de medir y analizar, tienen una confiabilidad del 100%, repetitivos y automatizables. Estos marcadores se encuentran generalmente en regiones no codificantes del genoma y distribuidos uniformemente (Goldstein y Schlotterer, 1999). Polimorfismo de Longitud de Fragmentos de Restricción (RFLP) Es una de las primeras técnicas descritas (Botsein et al., 1980), desde la aparición de la PCR, y consiste en visualizar las diferencias al nivel de la estructura del ADN, basándose en el uso de enzimas de restricción que cortan el ADN en sitios donde se encuentran una secuencia específica de nucleótidos. La identificación de los fragmentos (RFLPs) requiere del uso de geles de electroforesis que permitan separar los fragmentos que difieren en tamaño. La limitación de esta técnica es que únicamente identifica dos alelos por locus, por lo que la variabilidad obtenida es reducida. Otra limitación es que la aproximación con RFLPs, para la búsqueda de polimorfismo en productos de PCR no es metodológicamente efectiva al 100%, ya que muchos SNPs potenciales podrían no cambiar un sitio de restricción, y por tanto no serían detectados por ella (Vance y Othmane, 1998). Polimorfismo Amplificado Aleatoriamente (RAPD) Representan a marcadores que se basan en el uso de oligonucleótidos (iniciadores) cortos, los cuáles a través de una reacción PCR, que se caracteriza por temperaturas de «annealing» bajas, lo cual genera una serie de fragmentos de amplio espectro a partir del ADN molde. La habilidad consiste entonces en encontrar aquellos fragmentos amplificados y que resulten ser polimórficos, los cuáles pueden posterior-mente ser mapeados. Una de las principales limitaciones del uso de los RAPDs es la baja repetibilidad de los análisis, aunado a la necesidad de usar un gran panel de RAPDs, lo que representa un elevado valor económico y un laborioso trabajo analítico. Asimismo, y dado que es un marcador genético de tipo dominante, no podemos discriminar la existencia de heterocigotos, subestimando la cantidad de polimorfismo existente (Levin et al., 1994). Marcadores microsatélites
Dado que los microsatélites están más o menos distribuidos a lo largo de todo el genoma de los eucariotas, aunque con baja frecuencia en las regiones codificantes y, quizás también en los telómeros, su presencia en estas regiones se ha descrito asociada a enfermedades (Armour et al., 1994; Hancock, 1999; Tautz y Schlotterer, 1994). Sin embargo, aún se desconoce el significado funcional de estas secuencias, a pesar de que la hipótesis más aceptada apunta a que pueden estar relacionados con el empaquetamiento y la condensación del ADN en los cromosomas (Vanhala et al., 1998). Debido a particulares ventajas, el uso de microsatélites ha tenido un gran impacto en el estudio de la genética de animales y plantas. Desde su descubrimiento en 1989 (Litt y Luty, 1989, Tautz, 1989, Weber y May, 1989). El análisis con microsatélites involucra la detección de fragmentos específicos de ADN y nos da la medida de los alelos (en pares de bases, pb) en cada una de las regiones. Aplicaciones de los microsatélites Identificación Individual y Pruebas de Paternidades: El principio de las pruebas de paternidad utilizando marcadores genéticos consiste en la comparación del genotipo y/o fenotipo de la descendencia con el de sus progenitores. Como principio «mendeliano», uno de los alelos que presenta un individuo proviene del padre y el otro de la madre. En dicho análisis, al igual que la identificación individual, la identificación del (os) testigo (s), tanto a nivel fenotípico como genotípico debe ser perfectamente conocida y concordante con la información obtenida en los análisis realizados al individuo. El elevado polimorfismo que presentan estos marcadores y la posibilidad de poder detectar ambos alelos, los hace muy útiles para identificaciones individuales, ya que resulta muy poco probable que dos individuos elegidos al azar, si son analizados para una serie de marcadores, compartan todos sus alelos. Para seleccionar los marcadores a utilizar, estos deberían reunir las características descritas anteriormente y presentar además: alta variabilidad, herencia estable (baja tasa de mutación), elevada reproductividad y precisión, la no presencia de alelos «nulos», ser un procedimiento fácil, rápido, económico, potencialmente automatizable, que la información del genotipo pueda ser transferida rápidamente, que la fuente de ADN no esté limitada únicamente a muestras sanguíneas frescas ni a grandes cantidades de ADN y por último, presente una segregación independiente con otros marcadores al ser combinados en la prueba. Durante las últimas décadas las pruebas de paternidad en équidos (específicamente en caballos) se han venido realizando principalmente a través de la tipificación sanguínea, incluyendo tanto pruebas serológicas como grupos sanguíneos (hemotipados); así como, análisis electroforéticos del polimorfismo de proteínas y enzimas sanguíneas (alozimas). Siete sistemas de grupos sanguíneos y 16 polimorfismos bioquímicos han sido reconocidos internacionalmente y utilizados rutinariamente a nivel mundial, como herramientas oficiales para la prueba de paternidad (ISAG, International Society of Animal Genetics). La combinación de estos sistemas proporciona un 97% de probabilidad de detectar o asignar un padre o una madre incorrecta y cerca del 100% de probabilidad para un cruzamiento entre individuos de otras razas (Bowling y Clark, 1985, Bowling, 2001). Actualmente, con un conjunto aproximado de 10-12 marcadores microsatélites, se obtiene una efectividad teórica para detectar parentescos incorrectos (PE) por el orden del 99.99% (Aranguren-Méndez et al., 2001; Bowling et al., 1997). Mapas Genéticos y Genómica Comparativa: Otra aplicación, de los microsatélites es la construcción de mapas de ligamiento más completos y detallados; así como la identificación de genes de interés (QTL´s). Todos los marcadores pueden ser utilizados para mapas de ligamiento; sin embargo, se requiere, que los alelos se segreguen independientemente y que además puedan ser monitoreados a través del pedigrí. La descendencia puede ser informativa si los progenitores son doble heterocigotos en los loci analizados. Los loci situados en cromosomas distintos podrían recombinarse libremente durante la gametogénesis parental hasta un 50% (segregación independiente); mientras que, si se encuentran en el mismo cromosoma recombinarían con una frecuencia que oscila entre 0 a 50% dependiendo de la distancia en centi-morgans (cM) presente entre ellos. Así, un mapa genético bien surtido de marcadores se convierte en una herramienta muy útil para identificar genes responsables de caracteres de interés. Se trata de buscar asociación entre varios alelos, en cualquiera de los marcadores, segregando en poblaciones que presentan el carácter de interés, para identificar regiones del genoma donde es más probable que se encuentre el gen responsable de ese carácter (Cheng et al., 1995). Actualmente existen proyectos internacionales para elaborar mapas genéticos en las principales especies domésticas (Roslin Institute, http:// www.ri.bbsrc.ac.uk), los cuales describen abundantes marcadores de este tipo. Estudios de Genética Poblacional: Representa una de las áreas en donde los microsatélites han sido más ampliamente utilizados, ya que nos permite estimar los niveles de variabilidad genética dentro de las poblaciones y analizar las relaciones genéticas existentes entre las mismas. Este tipo de estudio es de gran importancia para realizar estimaciones de la diversidad genética y de la consanguinidad existente en poblaciones de animales domésticos en peligro de extinción. Durante los últimos años se han realizado muchos estudios que han utilizado los microsatélites para análisis filogenéticos, concluyendo que, con un buen número de loci analizados y con apropiadas tasas de mutación, los microsatélites pueden dar una buena aproximación de la filogenia (Aranguren-Méndez et al., 2001, 2002(a), 2002(b), 2002(c), 2002(d); Farid et al., 2000; Ishida et al., 1994; Ishida et al., 1994; Loftus et al., 1994; Peelman et al., 1998; Saitbekova et al., 1998; Takezaki y Nei, 1996). En los estudios de genética de poblaciones, los marcadores permiten la identificación de cada alelo por locus, la obtención de datos poblacionales, y el cálculo de las frecuencias alélicas. De esta manera podemos estimar las distancias genéticas entre poblaciones o entre individuos (Bowcock et al., 1995, Ponsuksili et al., 1999); así como también realizar análisis filogenéticos y de estructura de la población. La diversidad o variabilidad genética se puede definir como «la capacidad genética para variar», y por ende, la capacidad a responder tanto a variaciones de índole ambiental como a cambios en los objetivos de selección. Es así, como la variabilidad genética constituye la base del progreso genético (Rochambeau et al., 2000). Asignación de Individuos a Raza: Diferentes procedimientos han sido informados para este propósito, indicando una gran variedad de aplicaciones y de metodologías para la correcta identificación de la fuente poblacional (Cornuet et al., 1999, Paetkau et al., 1995, Rannala y Mountain, 1997). Los dos métodos más utilizados para asignar individuos a poblaciones o razas son: basados en probabilidades y basados en distancias genéticas. En los primeros, los individuos son asignados a aquella población en la que su genotipo presenta una mayor probabilidad de pertenencia; mientras que, en los segundos, los individuos son asignados a la población que genéticamente sea más cercana. Métodos Basados en Probabilidades: a.- Método de frecuencias: Este método fue descrito originalmente por Paetkau et al. (1995), y consiste en asignar un individuo a una población basándose en el valor de probabilidad de su genotipo individual de pertenecer a ella. La asignación por este método se realiza en tres etapas: a.1.-Calcula las frecuencias alélicas de todas las poblaciones potenciales. a.2.- Calcula la probabilidad de ocurrencia de cada genotipo multilocus para cada una de las poblaciones y a.3.- Asigna el individuo a aquella población en la cuál el genotipo multilocus obtuvo la mayor probabilidad. b.- Método Bayesiano: Este método es bastante similar al anterior y está muy relacionado con lo reportado por Rannala y Mountain (1997), quienes utilizaron la metodología Bayesiana para detectar migrantes mediante el uso del genotipado multilocus. Asumiendo una función de densidad igual a priori de las frecuencias alélicas de cada locus en cada población, Rannala y Mountain (1997), mostraron que la probabilidad marginal de observar un individuo con un genotipo AkAk en el locus j y en la población i es igual a: ((nijk+ 1 / Kj+ 1) (nijk+ 1 / Kj)) / ((nij+ 2) (nij+ 1)) si k = k' ((nijk+ 1 / Kj) (nijk' + 1 / Kj)) / ((nij+ 2) (nij+ 1)) si k ¹ k' donde nijk es el número de alelos k muestreados en el locus j y en la población i (que no contiene al individuo a ser asignado), nij es el número de copias de genes muestreados en el locus j y en la población i y Kj es el número total de alelos observados del locus j en las poblaciones estudiadas. Métodos Basados en Distancias: Los métodos basados en distancias asignan el individuo a la población genéticamente más cercana o próxima. Para ello se requiere definir una distancia entre el individuo y la población. Se han definido numerosas distancias genéticas; sin embargo las podemos agrupar en dos grandes bloques: las distancias entre poblaciones (p.e. distancias de Nei; distancia de cuerda de Cavalli-Sforza y Edwards, etc.) y las distancias entre individuos (eje. distancia de alelos compartidos (DAS)) (Chakraborty y Jin, 1993). En el primer caso se adaptan estas distancias a la relación entre individuo-población, considerando a los individuos cómo una muestra de dos genes (los posibles valores de frecuencias alélicas son 0, 0,5 y 1). Mientras que, en el caso de los alelos compartidos (DAS), la distancia entre un individuo y la población se toma como el promedio de distancia entre el individuo y los miembros de esa población (Aranguren-Méndez et al., 2002(b)). A pesar de las grandes ventajas que poseen los microsatélites como marcadores moleculares, existen también otros factores a considerar que pueden disminuir el poder y/o la sensibilidad de éstos como marcadores de elección, o pueden ser fuente de error que disminuya su utilidad en los estudios genéticos. Estos factores son: el patrón de mutación, los alelos nulos y la homoplasia. Mutación, alelos nulos y homoplasia en los microsatelites
Mutación: Las mutaciones son alteraciones del material genético y en ellas se incluyen desde simples sustituciones de un solo nucleótido hasta las deleciones o inserciones de uno o más nucleótidos. La mayoría de las mutaciones en animales no conllevan a cambios en el fenotipo, ya que, ocurren en regiones nocodificantes (mutaciones silentes). De forma general, las regiones o secuencias codificantes muestran una baja tasa de mutación que se ve reflejada en la escasa variabilidad existente dentro de especies y el alto grado de conservación que presentan estas regiones entre especies (Eisen, 1999). El elevado grado de conservación de estas regiones se pueden explicar muchas veces por el hecho de que las mutaciones dentro de esta región son deletéreas, ya que causan la pérdida de una función importante y por lo tanto son eliminadas por selección purificadora. Entender el proceso mutacional de los microsatélites es esencial antes de inferir las relaciones existentes entre la variación observada y las distancias genéticas o la estructura de una población. Los microsatélites, a diferencia de otros marcadores, tales como proteínas o enzimas, presentan un patrón diferente de mutación, ya que en primer lugar, la mayoría de las mutaciones están involucradas por la ganancia o pérdida de una simple unidad de repetición (Weber y Wong, 1993), además de la propia presencia de homoplasia, la cuál a su vez causa una subestimación de la cantidad total de variación entre poblaciones y por ende de las distancias genéticas; por lo tanto, ocurre una sobreestimación de las similitudes entre las poblaciones. La tasa de mutación en los microsatélites ha sido estimada en un rango que oscila entre 10-3 y 10-5 mutaciones por gameto (Bowcock et al., 1995, Forbes et al., 1995); sin embargo, el mecanismo cómo los microsatélites mutan es aún desconocido. Dos mecanismos son los que principalmente han sido propuestos: en primer lugar se cita un desigual cruce en la meiosis y en segundo lugar un desliz de la hebra del ADN durante la replicación, siendo al parecer esta última, la principal causa de mutación de los microsatélites (Goldstein y Schlotterer, 1999). In vitro, algunos factores intrínsecos, tales como, la longitud de la repetición y la composición (tipo de base nitrogenada) han demostrado que pueden afectar la tasa de mutación de los microsatélites. Es así como los dinucleótidos presentan tasas de mutación más elevadas que los trinucleótidos, y las secuencias con alto grado de AT (adenina-timina) en su composición mutan a mayores tasas que las que presentan altas combinaciones de GC (guanina-citosina) (Schlotterer y Tautz, 1992). Los dos principales modelos que se han utilizado para modelar el proceso mutacional de los microsatélites son: el modelo de alelos infinitos (IAM) y el modelo mutacional por pasos (SMM). En el IAM se supone que la mayoría de nuevas mutaciones dan lugar a nuevos alelos distinguibles, es decir, los nuevos alelos mutantes son siempre diferentes a los que ya existían en la población original, por lo que en este caso la homoplasia no existe o es despreciable. En el SMM los alelos sólo pueden mutar por la ganancia o pérdida de una sola unidad de repetición; debido a esto, queda claro que puede existir una gran cantidad de homoplasia. Ambos modelos son por lo tanto extremos. Otro modelo intermedio que se cita en la literatura, aunque con menor frecuencia es el de dos fases (TPM) y es igual al SMM, pero permite que se puedan dar mutaciones de mayor magnitud (Teoría de la Coalescencia) y predice la varianza esperada en número de repeticiones de un microsatélite, bajo distintos procesos mutacionales e historias demográficas. En este modelo se pueden dar «mutaciones viejas» (alelos ya existentes en la población), pero también «mutaciones nuevas» (alelos nuevos en la población), con lo cuál la cantidad de homoplasia existente siempre será menor que en el modelo SMM. Por lo tanto TPM en la práctica sería más parecido al IAM (Di Rienzo et al., 1994; Murray, 1996). Al principio se esperó que los microsatélites siguieran el SMM, pero los análisis experimentales han demostrado que no se trataba de un modelo simple (Kwok et al., 1996). Parece ser que, dependiendo del tamaño de la unidad de repetición, un microsatélite se adapta más a un modelo o al otro. Los microsatélites de repeticiones de 3-5 pb parece que sí siguen el SMM (Tautz y Schlotterer, 1994), mientras que los de 1-2 pb siguen el IAM o el TPM (Vanhala et al., 1998), al igual que los minisatélites (repeticiones de 10-60 pb) (Estoup et al., 1999). Alelos Nulos: Se habla de alelos nulos cuando estos no pueden ser amplificados por PCR, debido principalmente a una mutación en el punto de hibridación del iniciador. Uno de los alelos no amplifica y por lo tanto el individuo es catalogado como homocigoto para el otro alelo (Dawson et al., 1997). La existencia de alelos nulos es especialmente difícil de detectar cuando su frecuencia en la población es baja y cuando no se dispone de información genealógica fiable. Su determinación sería posible si se presentara en homocigosis, ya que no obtendríamos producto amplificado de un determinado individuo para ese locus. En los casos de verificación de paternidades, podemos sospechar la presencia de alelos nulos en un marcador cuando todos los demás marcadores apuntan a un progenitor y sin embargo, es homocigoto para ese marcador excluyente, siendo el descendiente homocigoto para ese marcador para uno de los alelos parentales. Un alelo nulo podría llevarnos a interpretaciones erróneas y excluir a estos animales como posibles progenitores en un análisis de paternidad, además de sesgar las estimaciones de las frecuencias alélicas en las poblaciones. Otra manera para detectar la presencia de alelos nulos sería a partir del cálculo del déficit de heterocigotos para el equilibrio Hardy-Weinberg (Neuman y Wetton, 1996). Para solucionar el problema de los alelos nulos se pueden diseñar cebadores alternativos fuera del pun-to de mutación y volver a analizar los individuos clasificados como homocigotos. Asimismo, esto se puede evitar no utilizando marcadores que han sido reportados como portadores de alelos nulos en ciertas poblaciones o razas, ya que pueden darnos problemas de esta índole (Dawson et al., 1997, Mundy y Woodruff, 1996). Homoplasia: Originalmente, el término «homoplasia» fue utilizado por los evolucionistas para referirse al hecho de que un mismo carácter, presente en dos especies, no siempre ha derivado del mismo carácter ancestral. A nivel genético, se dice que dos alelos son homoplásicos cuando poseen un estado idéntico, aunque no sea por descendencia (Estoup y Cornuet, 1999). La homoplasia se refiere al hecho de que dos alelos tengan el mismo tamaño (pb) pero no es debida a que sean idénticos. Se toma como idénticos por tener el mismo tamaño pero intrínsecamente existen claras diferencias en cuanto a su estructura, presencia de inserciones y/o deleciones, cambios de bases o variaciones en la región flanqueante (Primmer y Ellergren, 1998). Éste es un tipo de polimorfismo que puede detectarse únicamente por secuenciación, y puede pasar inadvertido en caso de analizar individuos mediante amplificación por PCR y electroforesis para asignar tamaños. La homoplasia en las pruebas de paternidad y parentesco nos supone una relación cuando no la hay o viceversa. También puede ser fuente de error en estudios poblacionales o de evolución porque la tasa de mutación de los microsatélites está relacionada de forma compleja con el tamaño y composición del alelo. Por ejemplo, los alelos interrumpidos o compuestos conllevan con más frecuencia a casos de homoplasia (Tautz y Schlotterer, 1994). En modelos mutacionales del tipo IAM se supone la no presencia de homoplasia, ya que una nueva mutación produce la creación de un nuevo alelo; en este caso totalmente distinto al que existía previa-mente en la población. Sin embargo, los otros modelos de mutación (SMM, TMP) pueden llegar a generar homoplasia y la cantidad dependerá de la tasa de mutación presente en la población. En los marcadores del tipo microsatélite se espera en teoría una cierta cantidad de homoplasia, ya que existe evidencia de que directa o indirectamente los microsatélites presentan un tipo de mutación del tipo SMM o TPM; los microsatélites se caracterizan por presentar una elevada tasa de mutación que puede oscilar entre 10-3 y 10 -5 mutaciones por locus y generación (Renwick et al., 2001); por último, el rango limitado de tamaños de alelos presentes en los microsatélites reduce los posibles estados alélicos favoreciendo la cantidad de homoplasia (Nauta y Weissing, 1996; Estoup et al., 1999). Análisis de la variabilidad genética Existe una gran diversidad de estadísticas para cuantificar la variabilidad genética y resumir la información a términos más manejables. Los estadísticos más empleados son: porcentaje de loci polimórficos, el número medio de alelos por locus, la heterocigosidad esperada (HE) y observada (HO) y el índice de contenido de polimórfico (PIC) (ArangurenMéndez et al., 2001, 2002(b). Porcentaje de loci polimórficos: Un locus se considera polimórfico si podemos detectar más de un alelo en una población. General-mente el criterio más utilizado es el del 1%, es decir, un locus será polimórfico si el alelo más común presenta una frecuencia menor al 99% en la población bajo estudio. Número medio de alelos por locus: Esta estadística indica el número medio de alelos que presenta un locus en una población. Sin embargo, dicha medida depende mucho del número de individuos analizados, ya que cuando el número es grande, mayor es la probabilidad de detectar alelos suplementarios. No obstante, este estadístico es útil para estudiar la existencia de variabilidad críptica en los loci. Heterocigosidad (H): Representa una mejor medida de la variación genética, ya que es precisa y no arbitraria. La heterocigosidad la podemos estudiar como HO y HE. La HO se define como la frecuencia relativa de individuos heterocigotos observados en la muestra para cualquiera de los loci y se calcula por cómputo directo. Mientras que la HE, desde el punto de vista matemático, es la probabilidad de que dos alelos tomados al azar de la población sean diferentes (Crow y Kimura, 1970). En una población en equilibrio H-W, la frecuencia de los heterocigotos viene dada por la ecuación 2 pq. El cálculo de la HE en la población puede realizarse a través de: HE= 1 - S pi 2 siendo pi2 = (homocigosidad) o también su equivalente: HE= S pi (1 - pi) siendo este termino también conocido como diversidad génica de Nei (1977). Según Zapata (1987), la HE es un buen estimador de la variabilidad, dado que se aplica a cualquier especie, independientemente de su estructura reproductiva o genética, pudiéndose por tanto realizar comparaciones entre ellas. Índice de Contenido Polimórfico (PIC): El PIC es similar al valor de heterocigosidad y oscila entre 0 y 1. Este índice evalúa la informatividad de un marcador en la población de acuerdo a las frecuencias de los alelos. Para su cálculo se multiplica la probabilidad de cada posible cruzamiento (estimado a partir de las frecuencias alélicas) por la probabilidad que sean informativos, es decir, que se pueda identificar al progenitor del que procede el alelo (Botsein et al., 1980). Donde pi...pn son las frecuencias de los n alelos. Probabilidad de exclusión (PE): Expresa la probabilidad de que la asignación errónea de un progenitor sea detectada por el análisis (Jamieson, 1994).
donde i > j y pi..pn son las frecuencias de los alelos. PEtot = 1 - ((1-PE1) (1-PE2)........(1-PEn)); a la probabilidad global de un conjunto de marcadores. Equilibrio Hardy-Weinberg (H-W): La ley de Hardy-Weinberg (Hardy, 1908; Weinberg, 1908) representa a una población grande de individuos diploides, con reproducción sexual aleatoria, sin selección, mutación y migración, las frecuencias génicas y genotípicas permanecen constantes de generación en generación y, además, existe una relación simple entre ambas. Así, una población con frecuencias génicas y genotípicas constantes, se dice que está en equilibrio H-W. Cuando la población se desvía de manera significativa de estas proporciones se habla de desequilibrio H-W, y se puede medir mediante el índice de fijación F (Wright, 1965), el cuál se expresa para un locus cualquiera como: F = (HE- HO) / (HE); Siendo HE y HO la heterocigosidad esperada y observada para ese locus, respectivamente. Cuando el índice de fijación F es igual a cero se indica que la población está en equilibrio; mientras que si F es diferente de cero, ya sea en forma positiva o negativa, indicaría que existe un déficit o exceso de heterocigotos, respectivamente. Análisis de la estructura poblacional Antes de realizar los análisis de la estructura poblacional es esencial probar la variación genética encontrada mediante los microsatélites, con el objeto de que no se haya violado alguna de los supuestos básicos en un análisis poblacional. Estos supuestos son tres: a) selectiva neutralidad de cada locus, b) la inexistencia de alelos nulos y c) independencia de los loci (Murray, 1996). Efectos como la selección, puede confundir los resultados, y cualquier loci que esté presumiblemente ligado a un carácter de interés selectivo debe ser descartado del análisis. Aunque la mayoría de los microsatélites se comportan de manera neutral (Murray, 1996). Para los estudios de la estructura de la población los análisis de la F-estadística han mostrado ser una herramienta útil para dilucidar los patrones y extraer la variación genética residente entre y dentro de las poblaciones. Según estos estadísticos, la variabilidad de una población global puede ser subdivida entre sus subpoblaciones (Aranguren-Méndez et al., 2002(d). Wrigth (1965) definió tres F-estadísticas: FIS, FIT y FST, como correlaciones entre unidades gaméticas en la población a diferentes niveles. Los estadísticos FIS y FIT explican las correlaciones entre dos unidades gaméticas tomadas al azar de una subpoblación y del total de la población como un todo respectivamente; y nos indica el exceso o déficit de heterocigotos presentes; mientras que el FST es la correlación entre dos gametos tomados al azar de cada una de las subpoblaciones y mide el grado de diferenciación genética entre dos subpoblaciones. Estos parámetros se relacionan a través de la siguiente expresión: (1 - FIT) = (1 - FIS) · (1 - FST) (1) Pudiéndose inclusive en algunos casos extender el análisis a niveles jerárquicos adicionales dentro de la población, y en casos de análisis de variabilidad genética intraracial, subdividiendo las razas en subpoblaciones, hasta dos niveles jerárquicos, por lo que la ecuación (1) extendida sería: (1 - FIT) = (1 - FIS) · (1 - FSC) · (1 - FCT) donde FSC y FCT miden el grado de diferenciación genética entre subpoblaciones dentro de razas y dentro de las subpoblaciones, respectivamente (5). La desviación del equilibrio H-W puede ser debida a una variedad de causas. Si los resultados arrojan un exceso de heterocigotos, podría en este caso ser atribuida a la presencia de una selección sobredominante o la ocurrencia de migraciones en la población estudiada; si por el contrario los hallazgos son de déficit de heterocigotos, podría deberse a cuatro factores principalmente; en primer lugar, que el locus esté bajo selección; en segundo lugar, a la presencia de alelos nulos en esa población, dando una falsa lectura de un exceso de homocigotos; en tercer lugar a altos niveles de consanguinidad en la población, producto de apareamiento entre individuos emparentados y, en cuarto lugar, se atribuiría a la presencia de una cierta subestructuración reproductiva de la población (efecto «Wahlund») (Nei, 1987). Nei (1977), reformuló los índices F, basándose en los valores de HO y HE. En este modelo, el cuál es independiente del número de alelos presentes en cada locus, la diferenciación o estructura de la población se mide por un parámetro denominado GST (Nei, 1973, 1977), análogo al FST y que cumple la siguiente relación: donde: HS es equivalente a la anterior HE, mientras que HT se define como la heterocigosidad media esperada por locus en la población total: HT= (åk hT) / k; siendo k el número de loci y hT la heterocigosidad esperada en la población total para un determinado locus. Una vez introducida la definición de diversidad genética de Nei (GST), HS viene a representar la diversidad genética dentro de la subpoblación y HT la diversidad genética en la población total. Análisis de la diferenciación genética Para estudiar las diferencias entre poblaciones se han de comparar las frecuencias alélicas observadas de cada locus mediante una prueba adecuada y calcular las distancias genéticas entre ellas. En este sentido, la distancia genética mide el grado de diferenciación existente entre poblaciones de una misma especie, o entre especies, pudiéndose cuantificar esa diferencia mediante diferentes índices de distancias. Las distancias genéticas ayudan a entender las relaciones evolutivas entre poblaciones y nos permite obtener información para caracterización de razas (Nagamine y Higuchi, 2001). De las distancias genéticas es importante estudiar el tipo de distancia, el método de construcción del dendrograma o algoritmos y el análisis de remuestreos. Distancias Genéticas: Las distancias genéticas son estimadoras del tiempo de separación entre poblaciones y se basan en ciertas diferencias genéticas existentes dentro y entre poblaciones. Algunas de las distancias genéticas están enfocadas en los cambios de las frecuencias génicas (p.e. FST); mientras que otras incorporan además el proceso mutacional, y en el caso específico de los microsatélites éstas se denominan distancias por paso o etapas (stepwise). Las matrices de las distancias genéticas entre poblaciones pueden ser convertidas en árboles evolutivos utilizando métodos de agrupamiento, tales como, el método de agrupamiento no ponderado por pares utilizando la media aritmética (UPGMA) o el Neighbour Joining (NJ) (Goldstein y Schlotterer, 1999). Suponiendo diferentes modelos mutacionales para los loci que codifican para proteínas (IAM) y para los loci microsatélites (SMM), Takezaki y Nei (1996) estudiaron la eficacia mediante simulación por ordenador de diferentes medidas de distancia en la correcta reconstrucción de una filogenia conocida. Dichas distancias correspondieron a: a.- Distancia estándar de Nei (DS (Nei, 1972)): DS = -ln (Jxy / Ö Jx Jy ) donde Jx y Jy son las homocigosis medias sobre loci en la población X e Y, respectivamente . Bajo el proceso mutacional IAM se espera que la DS incremente linealmente con el tiempo, si se mantiene un balance entre deriva y mutación a través de todo el proceso evolutivo. b- Distancia mínima de Nei (Dm (Nei, 1973) ): Dm = (Jx + Jy) / (2 - Jxy) En este caso la esperanza de Dmes conocida como: E(Dm) = J( 1 - e -2vt ), donde J es la homocigosis de las dos poblaciones. c.- Distancia de Reynolds (Reynolds et al., 1983): DL = -ln (1 - FST); FST @ qST = ((Jx + Jy) / (2 - Jxy)) / 1 - Jxy d.- Distancia de Rogers (Rogers, 1972):
e.- Distancia de Prevosti (Cp; (Prevosti et al., 1975) ): Esta distancia posee las mismas propiedades estadísticas que la DR y se define como:
f.- Distancia de cuerda (DC; (Cavalli-Sforza y Edwards, 1967) ): r mj DC = ( 2 p r) å Ö 2 (1 - å Ö xij y ij)
g.- Distancia «A» de Nei (DA; (Nei et al., 1983) ): Nei simuló esta distancia (DA) con el proceso mutacional IAM y demostró que era más eficiente que la DS, Dm, DR y DC en la asignación de correctas topologías. Otra distancia es la dm2 de Goldstein et al. (1995), la cuál es la más reciente y como producto de la aparición de los primeros estudios con ADN microsatélite. Dado que estos marcadores poseen un elevado grado de polimorfismo con respecto al número de repeticiones, su número puede aumentar o disminuir por un proceso mutacional del tipo SMM. Bajo este modelo de mutación, un alelo i (alelos con i repeticiones) se supone que puede mutar a otro alelo en un estado i + 1 ó i –1 con igual probabilidad, aludiendo un mejor análisis para este tipo de marcador y denotándose de la siguiente manera: donde mxj= (åi ixij) y myj= (åi iyij) representan los estados alélicos medios del j-ésimo locus y xij e yij son las frecuencias de los alelos en el estado i en el j-ésimo locus en la población X e Y, respectivamente. En estos estudios Takezaki y Nei (1996) concluyen con respecto a las distancias genéticas generadas a partir de datos de estudios con marcadores clásicos, los cuáles presentan un modelo de mutación IAM, y con microsatélites con mutación del tipo SMM. Las distancias más eficientes para ambos modelos resultaron ser DC y DA para la correcta asignación de topologías, y la distancia estándar (DS) y dm2 resultaron ser las más apropiadas para estimar los tiempos evolutivos, coincidiendo con otros reportes (Aranguren-Méndez et al., 2002(b), Nagamine y Higuchi, 2001, Ruane, 1999). Matemáticamente, cualquiera de estas funciones debe poseer ciertas propiedades para poder ser consideradas como una distancia. Primero, la distancia entre una población A y ella misma debe ser cero (0), de aquí que d(A,A)=0. Segundo, la distancia entre la población A y la población B debe ser simétrica, o lo que es lo mismo d(A,B) = d(B,A); si una distancia satisface estas propiedades se denomina distancia semimétrica y si además logra satisfacer la desigualdad triangular d(A,B) d» (d(A,C) + d(B,C)) se denomina distancia métrica (Eding y Laval, 1999); la distancia DS de Nei, no cumple estas propiedades, ya que no es una distancia métrica. Métodos de Agrupamiento o Algoritmos: En la construcción de los árboles filogenéticos existen diferentes métodos para dibujar la matriz de distancias. Sin embargo, los más empleados son el NJ y el UPGMA, los cuáles generalmente dan muy buenos resultados. A pesar de ello el primero parece ser superior cuando se suponen en el modelo diferentes tasas de evolución (Eding y Laval, 1999, Takezaki y Nei, 1996). El método NJ fue descrito por Saitou y Nei (1987), se caracteriza por que construye árboles mediante sucesivos agrupamientos de alineaciones, tomando en cuenta las longitudes de rama para dichos agrupamientos. Una vez realizado el agrupamiento deriva la mejor aproximación para la máxima parsimonia, construyendo los grupos apareados sobre la base de la mínima longitud de rama. Las dos principales características de este método son que los resultados nos dan un árbol no rotado (sin raíz u origen evolutivo), y luego este no supone un reloj evolutivo. Por otra parte, el método UPGMA construye los árboles o dendrogramas mediante sucesivos agrupamientos bajo el criterio del promedio de la distancia de cada par. Se caracteriza porque supone una tasa constante de cambios evolutivos, es decir, supone la existencia de un reloj evolutivo y los árboles son rotados. Weir (1996) lo definió como una distancia entre grupos, producto del promedio de todas las distancias entre pares de cada una de las combinaciones. Es así como los dendrogramas no son más que la representación gráfica de una matriz de distancias, y que bajo ciertas condiciones, esta representación puede ser tomada como una estimación de la filogenia Análisis de Remuestreos: Dado que la estimación de las distancias genéticas entre poblaciones es un método estadístico, el proceso de muestreo es sumamente importante. Existen varias técnicas estadísticas para remuestreos de los datos de loci genéticos. Entre ellas se encuentran el «Bootstrapping» y el Jackknife. Estas técnicas permiten la posibilidad de dibujar múltiples árboles y estimar la fiabilidad para los diferentes nodos en el árbol, así como el nivel de confianza del árbol (Weir, 1990). El «bootstrapping» es una técnica que genera información adicional acerca de la distribución de los parámetros. Dado que los datos consisten en n observaciones, un «bootstrap» genera una muestra de n dibujos al azar, realizado por medio del reemplazamiento de los datos originales. De este modo, todas las observaciones originales tienen la misma oportunidad de ser reemplazados, y al final el valor de los nuevos «bootstrap» será el promedio para el parámetro muestreado (Tivang, 1994; Weir, 1990). Generalmente este proceso se repite un gran número de veces (³ 1000), creando un gran número de «nuevos» conjuntos de datos, pudiéndose usar además para estimar medias y desviaciones estándares de un estimador calculado con los datos en cuestión. La técnica del Jackknife, por otro lado, es un simple re-muestreo numérico, que consiste en generar un «nuevo» valor estimado a partir de la técnica iterativa basada en la estimación de máxima verosimilitud después de excluir la i-ésima observación. Así, cada nuevo valor estimado estaría basado en n-1 observaciones. La principal limitación de esta técnica es la poca información que proporciona acerca de la distribución de los estimadores (Weir, 1996). Diversidad de Weitzman: El análisis de Weitzman es otro método para la construcción de árboles filogenéticos basado en la función de diversidad (Weitzman, 1992, 1993). Dado que en los programas de conservación genética el principal objetivo es preservar la variabilidad dentro de la población, bajo la hipótesis de existencia de correlación entre la variación genética y la viabilidad de la población, se hace necesario cuantificar la biodiversidad para así racionalizar mejor la política de conservación (Weitzman, 1992). La diversidad de Weitzman se define como la diversidad esperada después de un cierto periodo de tiempo, y se basa en la probabilidad de extinción de cada elemento (población o raza) en un conjunto o grupo considerado. Si n elementos están en peligro de extinción, ambos patrones (supervivencia y/o extinción) pueden ocurrir con una cierta probabilidad. Para cada uno de ellos pueden calcularse los correspondientes valores de diversidad (Thaon D'arnoldi et al., 1998). Una vez conocida la consanguinidad de una población o raza, ésta puede utilizarse como su propia probabilidad de extinción, proporcionando un orden prioritario sobre las razas a conservar (Aranguren-Méndez et al., 2002(c)). CONCLUSIONES
Los análisis con marcadores genéticos del tipo microsatélite para el estudio de poblaciones resultan muy útiles y valiosos, ya que además de la evaluación de la variabilidad genética, la información generada permite estudiar con mayor exactitud la estructura genética y la caracterización molecular de las poblaciones y/o razas. Así mismo, esta revisión confirma que los microsatélites pueden ser usados eficazmente para establecer las relaciones genéticas entre poblaciones y razas, de gran utilidad en la evaluación global de la diversidad (dentro y entre razas), que es sumamente útil y debería ser tomada en cuenta a la hora de tomar decisiones para poner en práctica planes de conservación y mejoramiento animal. Finalmente, la información generada por los microsatélites puede ser integrada al Banco Global de Diversidad de los Animales Domésticos de la FAO (FAO Global Data Bank on Domestic Animal Diversity (DAD-IS)), para el conocimiento y divulgación de las razas y poblaciones en estudios biológicos. LITERATURA CITADA
© 2005 ALPA. Arch. Latinoam. Prod. Anim. The following images related to this document are available:Photo images[la05005t1.jpg] [la05005f1.jpg] [la05005f2.jpg] |
| |||||||||

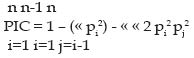
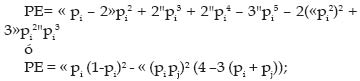
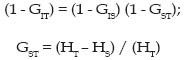
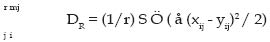
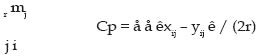
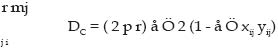
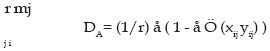
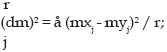
{kind=link}
{kind=link}