 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
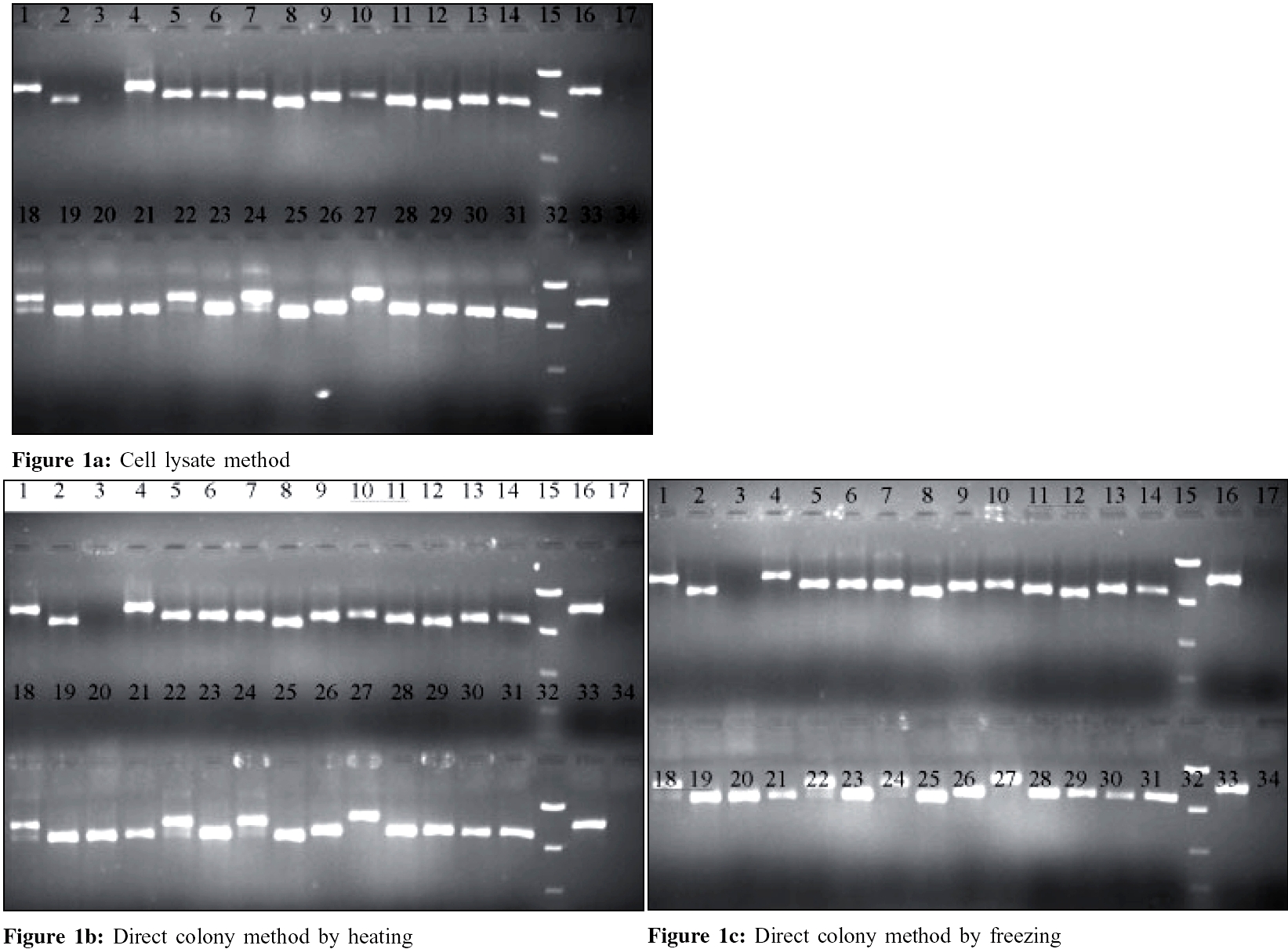
Indian Journal of Medical Microbiology, Vol. 24, No. 2, April-June, 2006, pp. 127-130 Brief Communications Evaluation of simplified DNA extraction methods for EMM typing of group a streptococci Jose JJM, *Brahmadathan KN Department of Clinical Microbiology, Christian Medical College, Vellore - 632 004, Tamil Nadu Code Number: mb06036 Abstract Simplified methods of DNA extraction for amplification and sequencing for emm typing of group A streptococci (GAS) can save valuable time and cost in resource crunch situations. To evaluate this, we compared two methods of DNA extraction directly from colonies with the standard CDC cell lysate method for emm typing of 50 GAS strains isolated from children with pharyngitis and impetigo. For this, GAS colonies were transferred into two sets of PCR tubes. One set was preheated at 94°C for two minutes in the thermal cycler and cooled while the other set was frozen overnight at -20o C and then thawed before adding the PCR mix. For the cell lysate method, cells were treated with mutanolysin and hyaluronidase before heating at 100o C for 10 minutes and cooling immediately as recommended in the CDC method. All 50 strains could be typed by sequencing the hyper variable region of the emm gene after amplification. The quality of sequences and the emm types identified were also identical. Our study shows that the two simplified DNA extraction methods directly from colonies can conveniently be used for typing a large number of GAS strains easily in relatively short time.Keywords: Group A Streptococci (GAS), emm typing, direct colony PCR Group A streptococci (GAS) are responsible for human infections ranging from mild sore throat and impetigo to fatal conditions such as streptococcal toxic shock syndrome.[1] They are also responsible for sequelae such as rheumatic fever / rheumatic heart disease (RF/RHD) and glomerulonephritis which lead to considerable morbidity and mortality in developing countries like India.[2] Knowledge about the GAS types distributed in specific geographical areas is required for designing suitable vaccine(s) as well as to make policies for global eradication of these sequelae.[3] The typing of GAS is now based on sequencing the hyper variable region of M protein gene ( emm ) through specific polymerase chain reaction (PCR) products and is called emm typing.[4] PCR is a powerful technique that serves as the basis for application of other advanced techniques like sequencing. Therefore, it is important to provide quality nucleic acid template free of inhibitory substances for amplification.[5] This is important when PCR is performed directly on clinical samples or rapid results are required. But for typing bacteria using pure cultures, the isolation of DNA from rest of the cellular components is not very essential, if DNA could be extracted and inhibitory substance(s) denatured without damaging the DNA. The application of colony PCR was first used for the rapid characterization of transformed Escherichia coli and phage studies.[6],[7] Subsequently, DNA amplified directly from bacterial colony was sequenced to yield results as good as those obtained with DNA extracted by the conventional phenol-chloroform procedure.[8] In an effort to minimize the steps involved in DNA extraction for emm typing by sequencing, we compared the standard CDC cell lysate method with direct colony method of DNA preparation. Materials and Methods A total of 50 GAS strains isolated from throat cultures of asymptomatic children (n=33), those with pharyngitis (n=7) and skin lesions (n=10) seen during an epidemiological survey conducted during 2001-2002 in a rural school situated 12 Kms from Vellore town, were selected for this study. They were isolated and identified, using methods standardized in our laboratory.[9] All GAS strains were preserved by lyophilization at +4oC for further studies. Control strains The cell lysate method was done as recommended by the CDC method.[4] One loopful of culture grown overnight on 5% sheep blood agar was suspended in 300 µL of 0.85% of NaCl and heated for 15 minutes at 70°C. Cells were suspended in 50 µL TE buffer [(10 mM Tris, 1mM EDTA (PH 8) containing 300 U/mL of mutanolysin and 30 µg/ mL of hyaluronidase (Sigma-Aldrich, Bangalore)] and were allowed to react for 30 minutes at 37°C. Samples were then heated at 100°C for 10 minutes and immediately cooled in ice before adding into the master mix. For the direct colony method, few GAS colonies from a blood agar plate were applied on the bottom of two sets of PCR tubes using sterile tooth picks. One set was frozen overnight at -20°C and thawed while the other set was preheated at 94°C for 2 minutes in the thermal cycler and cooled. Finally, the master mix was added into the two sets of samples separately. The thermal cycling parameters and the PCR mix with emm specific primers were carried out as per the CDC protocol (http://www.cdc.gov/ncidod/biotech/strep/protocols.htm). A PCR mix of 20 µL was prepared for each reaction. For direct colony PCR, the template volume was replaced with water and the thermal cycling conditions with extra 10 minute denaturation time as the initial step. The amplified product was detected by electrophoresis using 1.5% agarose gel at 100 volts for 45 minutes along with a low range quantitative DNA ladder (Invitrogen, California, USA.) and controls. EMM typing by gene sequencing Results All 50 strains could be typed by sequencing the hyper variable region of the emm gene after amplification by the three methods, namely, the cell lysate method of CDC, direct colony PCR by heating (94°C for 2 minutes) and direct colony PCR by freezing. The quality of sequences and the emm types identified by the three methods were identical. Thirty emm types were identified among the 50 strains studied [Table - 1]. Type 122 accounted for 5 strains, followed by types 8, 49 (4 each), 1, 57 and 58 (3 each), 63, 71, 85 and st 2147 (2 each). These ten types accounted for 30 (60%) of the 50 strains tested. Two types, stKNB3 and stKNB4 have been recognized by the CDC as new types. Discussion Our study confirms that physical methods such as overnight freezing at - 20°C or heating at 94°C for 2 minutes can be conveniently used for denaturing the cell wall. The DNA released is sufficient enough for amplification and subsequent sequencing. Thus these methods can not only replace more cumbersome and time-consuming cell lysate method, they can be used for typing large number of strains in much less time. Availability of adequate sample of quality DNA template, free of any inhibitory substance through amplification, is one of the critical factors in PCR.[5] Unlike PCR for rapid detection of pathogens or uncultivable organisms from clinical samples, PCR with pure culture is more direct and contains less inhibitory substances. We observed that bacterial cells obtained by touching a single colony with a sterile tooth pick can yield sufficient DNA for amplification. Qualitatively and quantitatively the amplified product is sufficient enough for sequencing and yields typing results as good as those obtained from the cell lysate method. Therefore, it is advantageous to standardize these methods for easy and rapid extraction of DNA directly from isolated colonies for emm typing of GAS strains. Extraction of DNA from gram positive bacteria such as GAS involves multi-step procedures which sometimes may prove to be ineffective.[11] They were simplified by treating the cell wall with glycine, enzymes or with ionic detergents.[12] Further modification by treatment with mutanolysin and hyaluronidase as in the cell-lysis method[4] is now a widely accepted protocol for emm typing of GAS. However, this method could be cumbersome, time-consuming, involves multiple reagents and is open to mistakes when large number of strains is being typed. Our study shows that this problem can be avoided by extracting DNA from bacterial cells by inexpensive physical methods such as freezing or heating. The latter methods [Figure - 1] were also successful in identifying one of the strains which repeatedly gave double bands in the cell-lysis method, indicating the presence of non-specific primer binding region. Improper application of colonies or a heavy inoculum may result in inadequate amplification and therefore precaution needs to be taken to ensure application of optimum number of colonies for the DNA extraction. We also observed that GAS strain M18 (MGAS 8232) included as a control failed to amplify when 48 hour-old culture was used for the freezing or heating methods. Therefore, a 16-24 hour old culture is necessary for optimal DNA amplification.[13] The advantage of using physical methods such as heating and freezing/thawing as pretreatment methods is that they are simple, time saving and economical. In the former, DNA is ready for amplification immediately and is therefore more ideal than the latter method where the sample needs to be kept frozen overnight. This makes the work easier and allows handling large number of samples for emm typing in a short time. Acknowledgement We thank the Department of Clinical Haematology, CMC Vellore, for offering us the gene sequencing facility for emm typing and Dr. Nancy Hoe, Rocky Mountain Laboratory, Hamilton, Montana, USA for teaching one of us (JJMJ) the freezing technique of direct colony PCR.References
Copyright 2006 - Indian Journal of Medical Microbiology The following images related to this document are available:Photo images[mb06036t1.jpg] [mb06036f1.jpg] |
| |||||||||
{kind=link}
{kind=link}