 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
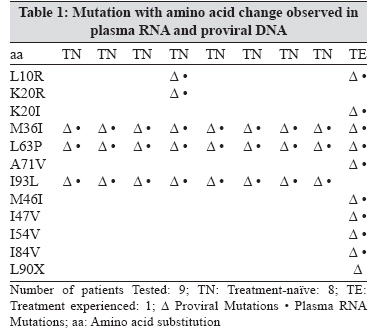
Indian Journal of Medical Microbiology, Vol. 26, No. 2, April-June, 2008, pp. 151-154 Brief Communication Genotypic resistance profile of HIV-1 protease gene: A preliminary report from Vellore, south India Kandathil AJ, Kannangai R, Abraham OC, Sudarsanam TD, Pulimood SA, Sridharan G Department of Clinical Virology, Christian Medical College, Vellore - 632 004, Tamil Nadu Date of Submission: 01-Mar-2007 Code Number: mb08043 Abstract HIV-1 subtypes other than B are responsible for most new HIV infections worldwide; virus sequence data for drug resistance is described only from a limited number of non-B subtype HIV-1. This study is on mutations and polymorphisms of HIV-1 protease gene that can predict drug resistance in subtype C. The genotypic resistance assay was carried out on 38 HIV-1 strains with their plasma RNA and in nine, the proviral protease gene was sequenced. The treatment naοve strains showed minor resistance mutations, there were no major resistance mutations in the protease gene. We suggest the use of resistance testing to monitor individuals on therapy and also before initiation of therapy, gathering more sequence information for a data bank of Indian strains.Keywords: Drug resistance, India, HIV-1, protease gene mutations A major obstacle to the long-term efficacy of antiretroviral therapy is the emergence of human immunodeficiency virus-1 (HIV-1) variants with reduced susceptibility to antiretroviral agents. [1] The frequency of one or more major resistance mutation in recently infected individuals increased from 3.8% to 10.2% while on treatment in a five- year period as reported from the West. [2] Mutations seen in the HIV-1 reverse transcriptase (RT) and protease region are classified as primary and secondary mutations. Primary mutations are mutations which reduce drug susceptibility directly whereas secondary mutations reduce drug susceptibility or improve replicative fitness of the virus in conjunction with primary mutation. [3] Although HIV-1 subtypes other than B are responsible for most new HIV infections worldwide, the understanding of drug resistance of HIV-1 is largely limited to those areas of the world where subtype B viruses predominate and antiretroviral therapy has been accessible. In the year 2000, non-B subtypes accounted for 88% of new HIV-1 infections, with subtype C responsible for 47.2%. [4] Hence, the present report is on findings of a study on drug resistance mutations and polymorphisms in the protease gene of HIV-1 that can predict drug resistance in subtype C, the virus that primarily circulates in India. Materials and Methods Blood samples were collected from 38 (35 treatment naÏve and 3 treatment failure) HIV infected individuals who had come to the Clinical Virology department of a tertiary care centre in India (south) for their CD4+ T cell estimation and/or HIV-1 viral load during the year 2004-2005. Informed consent was obtained from all the individuals before we proceeded with the test. Viral load estimation in all the individuals was done using Artus RealArt™ HIV 1 kits (Qiagen Hamburg GmbH, Germany) using Rotor-Gene™ 3000. CD4 estimation was done using Guava ® EasyCD4™ System (Guava Technologies, Hayward, CA, USA) as described earlier. [5] The genotypic resistance assay was carried out on 38 strains by sequencing the protease gene. The HIV-1 protease gene was amplified from extracts of plasma RNA using QIAamp ® viral RNA extraction kit (Qiagen GmbH, Hilden, Germany). Following cDNA synthesis, amplification of the protease gene was done using forward primer PT-1 ACCAGAGCCAACAGCCCCACCA and reverse primer CPT-2 CCTTTGGGCCATCCATTCCTGGC. [6] The cycling conditions were as follows: 95°C for 15 minutes for activation of the Hotstart Taq enzyme followed by 40 cycles of 95°C for 45 seconds, 60°C for 45 seconds and 72°C for a minute followed by a final extension at 72°C for 10 minutes. For nine random strains of the thirty eight samples, the HIV-1 protease gene was amplified also from proviral DNA extracted from peripheral blood mononuclear cells (PBMC). The DNA was extracted using QIAamp ® blood DNA extraction kit (Qiagen GmbH, Hilden, Germany). The cycling conditions were the same as the one followed for cDNA amplification. Products from the PCR were first analysed on an ethidium bromide stained agarose (2%) gel to check for the specific 330 bp size amplicon using the gel documentation system Geldoc 2000 (BioRad, California, USA) using the software Quantity One, version 4.1.1(BioRad, California, USA). The sequencing of the amplified products was done using Big Dye terminator assay on ABI PRISM 310 Genetic Analyzer (PE Applied Biosystems, California, USA). The sequencing was done at an institutional facility. The sequences were then submitted to the Stanford HIV Drug Resistance database for analysis (http://hivdb.stanford.edu). Results The average HIV-1 viral loads for the treatment-naÏve group was log 10 5.55 copies/mL, with a median value of log 10 4.93 copies/mL. In the treatment experienced group, the average load was log 10 6.38 copies/mL with a median value of log 10 5.32 copies/mL. The CD4 counts in the treatment-naοve patient ranged from 36-963 cell/mm 3 with a median value of 179 cells/mm. 3 The CD4 value for the treatment experienced group was available only for one patient (patient no. 3-CD4: 83 cells/mm 3 ). Based on results obtained from the Stanford data base all but one of the strains from treatment experienced group was identified to be as clade C. The one non-clade C strain was identified as clade A. All the 38 strains wherein the protease gene of plasma RNA was sequenced showed mutations in positions 63, 93. Thirty seven strains (97%) had an additional mutation at 36. Three strains (8%) showed mutations at position 20, two (5%) at 60 and one (3%) each at positions 10 and 77. In this study, we have analysed proviral DNA taken from PBMC for nine of our patients including from a patient who was showing treatment failure. The differences in the mutations observed in HIV-1 strains from the two different sources are shown in [Table - 1]. The mutations observed in the HIV-1 strains amplified from the treatment experienced patients are shown in [Table - 2]. Two of the treatment experienced patients showed a major mutation at position 46. All the three patients had mutations at position 35. Discussion All the strains from treatment-naοve individuals showed minor resistance mutations in the protease gene. There were no major resistance mutations in this group. There was no difference in the mutations observed when analyzing sequences obtained from proviral and plasma RNA in the treatment-naÏve group. There was an additional mutation observed in the proviral sequence of the patient belonging to the treatment experienced group. In the absence of any drug exposure, protease sequences from B and non-B HIV-1 are polymorphic among 30% of the protease gene coded amino acids. [7] Some of these amino acid substitutions may occur at high rates in non-subtype B viruses at positions associated with drug resistance in subtype B viz., 10, 20, 36, 63, 71, 77 and 93. [7] It is not clear whether this may reduce the activity of some protease inhibitors (PIs) or if this is a basis for the evolution of different primary resistance and cross-resistance pattern. [8] In our study, 38 strains showed mutations in positions 63, 93 and 37 strains had an additional mutation at 36. At this point of time we do not know which of these are likely to be responsible for treatment failure subsequently. Reduced susceptibility to more than one PI is most likely to be associated with amino acid substitutions at six positions: 10, 46, 54, 82, 84 and 90. [9] One of our patients had mutations at position 10. This in a treatment-naοve population is indeed disconcerting with portentous implications to rationalisation of HAART in India. In one sentinel study from India, wherein phenotypic analyses was carried out on treatment-naοve strains, primary resistance to protease inhibitors was 2.5%. [10] Based on data obtained by genotypic analyses of strains from India, 20% of the strains analysed showed mutations at sites conferring resistances to PIs. [6] In our study none of the strains from the treatment-naÏve group had any major protease resistance conferring mutation. The three treatment experienced individuals had different major mutations conferring resistance to PIs. This can be attributed to the fact that they were on different PIs. The duration of HAART in these individuals ranged from 48 to 84 months (62.6 ± 19). Patient 1 had the following major resistance mutations: D30N and M46I. The D30N and N88D are mutations selected during nelfinavir therapy with D30N appearing first. [9] The major protease mutations seen in patient 2 were M46I, I47V and I54V. I47V is reportedly found in lopinavir experienced patients and is also commonly selected during amprenavir therapy. [11] The M46I and I54V are mutations that accumulate during failure of therapy with PIs. Patient 3 showed two major resistance mutations namely V82I and L90M. L90M is a resistant mutation that is observed during failure of therapy with protease inhibitors. [11] Patient 2 and 3 also showed secondary mutation E35D. In this mutation which occurs at amino acid position 35 of the protease gene sequence, glutamic acid is replaced by aspartic acid. E35D mutation affects the conformational equilibrium between the closed and semi-open conformations of the free protease and also causes a significant reduction in its binding free energy of the protease for its substrate and amprenavir. It has also been shown that the E35D mutation reduces interaction with the HLA B44 molecule impeding cellular immune response. [12] It is thus postulated to favour escape from the immune system in addition to conferring drug resistance. [12] Plasma viruses are the material of choice for early detection of PI resistant mutations during antiretroviral therapy. [13] In this study, we have analysed proviral DNA taken from nine of our patients including from a patient who was showing treatment failure. It is suggested that strains with drug resistant mutations are frequently accompanied by loss of replication capacity. [14] There have also been clinical observations wherein the wild-type virus emerges again when patients with extensive drug resistance are taken off therapy. [15] The genotypic data for the strains from different regions of India including south, showed the predominant subtype to be clade C. [16] Hence, it would be appropriate to assume that most of our strains were non-B subtypes. This fact was further strengthened by data from the Stanford HIV drug resistance database. The Stanford HIV genotypic drug resistance database algorithm generates mutations based on a consensus clade B sequence. Hence, one possible drawback of analyzing non-subtype B strains against this database could be that certain mutation/polymorphism in non-clade B strains could be missed or exaggerated. Based on our findings, we suggest that the use of resistance testing should not be restricted to monitor individuals on therapy but should also be used before initiation of therapy for individualised decision making. We also suggest gathering as much sequence data as possible to facilitate building a data bank of Indian strains. This will help in the future HAART strategy when it becomes more accessible to infected individuals. References
Copyright 2008 - Indian Journal of Medical Microbiology The following images related to this document are available:Photo images[mb08043t1.jpg] [mb08043t2.jpg] |
| |||||||||
{kind=link}
{kind=link}