 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
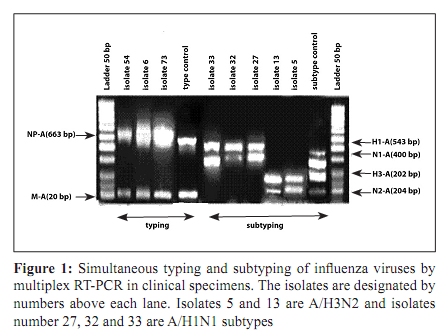
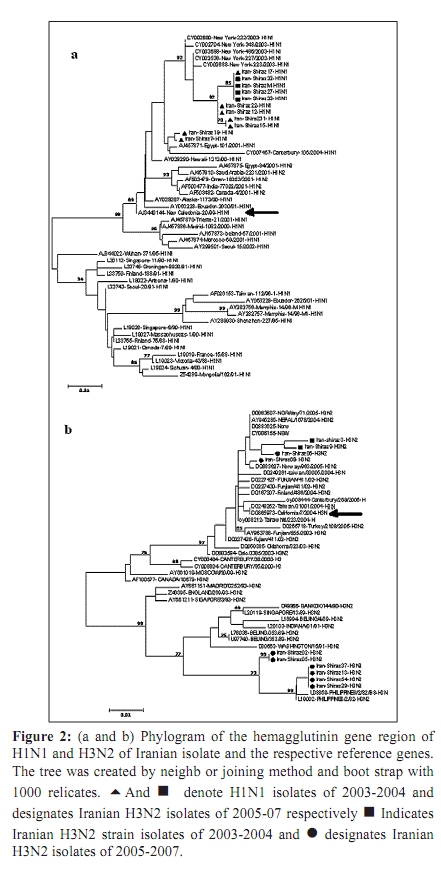
Indian Journal of Medical Microbiology, Vol. 28, No. 2, April-June, 2010, pp. 114-119 Original Article Antigenic variations of human influenza virus in Shiraz, Iran *A Moattari, H Ashrafi, MR Kadivar, MT Kheiri, M Shahidi, M Arabpour, A Ghanbari Virology Department, Shiraz University of Medical Science Shiraz-Iran (AM, HA, MA, AG), Pediatric Department, Namazee Hospital, Shiraz University of Medical Sciences Shiraz-Iran (MRK), Influenza Unit, Pasture Institute of Iran, Tehran, Iran (MTK, MS) Correspondence Address: *Virology Department, Shiraz University of Medical Science, Shiraz, Iran, moattaria@sums.ac.ir Date of Submission: 10-Jan-2009 Code Number: mb10036 PMID: 20404455 DOI: 10.4103/0255-0857.62486 Abstract Purpose: Influenza virus is a major cause of human respiratory infections and responsible for pandemics and regional outbreaks around the world. This investigation aims to determine the prevalent influenza genotypes during 2005-2007 outbreaks in Shiraz, the capital city of Fars province, southern Iran and compare the results obtained with those of previous study.Materials and Method: Of the 300 pharyngeal swabs collected from influenza patients, 26 were found to be positive by culture and hemagglutination (HA) assays. Typing and subtyping of the isolates carried out by using multiplex RT-PCR and phylogenetic analysis performed on isolated HA genes using neighbour-joining method. Result: Out of 26 positive isolates 12 and 14 were H1N1 and H3N2 respectively. The phylogenetic and amino acid sequence analyses of our H1N1 isolates showed 99-100% genetic resemblance to A/NewCaledonia/20/99 (H1N1) vaccine strain. Most of the Iranian H3N2 isolates varied form A/California/7/2004 vaccine strain in 20 amino acids of which positions 189,226 and 227 were located in antigenic sites of HA1 molecule. These substitutions were not observed in any of the H3N2 subtypes from the same region reported previously. Conclusion: The H3N2 subtype strains prevalent during the 2005/7 influenza outbreak in southern Iran demonstrated a drastic antigenic variation and differed from A/California/7/2004 vaccine strain. The H1N1 subtypes showed a notable resemblance to A/NewCaledonia/20/99 vaccine strain and therefore were predicted to be capable of conferring sufficient immunity against H1N1 subtypes. Keywords: Antigenic variation, human influenza virus, Iran, nucleotide sequence, phylogeny, Shiraz Introduction Influenza viruses are enveloped negative-stranded RNA viruses, which belong to orthomyxoviridae family. They are classified into three types - A, B and C, depending on antigenic differences in nucleoprotein and matrix protein. Further subdivision of these viruses into different subtypes is based on antigenic diversities of hemagglutinin (HA) and neuraminidase (NA) surface glycoproteins. [1],[2],[3],[4] Epidemics of influenza disease in humans are caused by influenza types A and B; infection with type C is subclinical. Worldwide, influenza virus infections are associated with significant morbidity and mortality. [5] New human subtypes of influenza viruses may emerge through drastic antigenic changes (antigenic shift) by genetic reassortment imposed by on major co-infection with influenza viruses from diverse animal species. [6],[7] The potential immunizing antigens of influenza viruses are HA and NA glycoproteins. Antibodies against HA molecule neutralize the virus and exert a highly selective pressure on viral infectivity. Influenza viruses are prone to high frequency mutations. Point mutations in HA and NA genes gradually evolve (antigenic drift) accumulate and leads to the emergence of immunologically distinct variants. [8] The cellular receptor binding sites and five hyper variable regions of globular head of the HA molecule are mainly the sites of antigenic variations. The efficacy of the vaccine is highly dependent on the extent of similarities between HA molecules of vaccine and epidemic strains. [9],[10] In this connection, we describe antigenic and genetic analysis of prevalent strains of influenza virus as an aid to the preparation of most effective vaccine. Materials and Methods Virus isolation Influenza A and B virus reference strains were obtained from National Institute for Biological Standards and Control (NIBSC) from the United Kingdom. The A/Newcaledonia/20/99H1N1, A/California/7/2004 H3N2 and B/shangdong/7/97 viruses were used as reference strains. Clinical samples (throat swabs) were collected within 72 hours of the onset of symptoms from 300 individuals aged from 1-68 years with influenza-like illness who referred to the out-patient clinics of Shiraz University of Medical sciences, Shiraz, Iran during 2005-7. The samples were transferred to virus transport medium, dispatched to the virology laboratory of medical school, and stored at -70 0 C until tested. The swabs were then vortexed in 5 milliliter medium for a few minutes to dislodge and suspend adherent viruses. The Madin Darby Canine Kidney (MDCK) confluent monolayer cells were inoculated with 200 micro litres of the viral suspension. The infected cell culture were maintained in serum free Dulbeco's Modified Eagle's (DMEM) (Sigma) supplemented with 2 mg/ml trypsin (Gibco BRL, life technologies) 100 µg/ml streptomycin and 100 unit/ml penicillin G. [11] The cultures were incubated at 34 0 C and examined daily for cytopathic effect which was confirmed by the ability of infected cultures to agglutinate guinea pig erythrocytes no later than seven days post infection. [11],[12] RNA Extraction and cDNA Synthesis RNA extraction was carried out using 300 µl of cell cultures inoculated with clinical specimens as well as reference strains and utilizing a commercial RNXplus TM solution (CinnaGene, Tehran, Iran). The viral RNA was eluted in 50 µl of DEPC treated water and immediately placed at -70 0 C. Super ScriptTM III First- Strand Synthesis Kit (Invitrogen, Carslbad, CA), was used for cDNA synthesis according to the manufacturer's instructions. [13] Primer and Multiplex RT-PCR As previously described, the differentiation between types A and B influenza viruses distinguishing H1N1 from the H3N2 subtypes were carried out based on the information on specific primers maintained in the Gene Bank. Primers utilizing the matrix protein gene of influenza A virus (M-A), nucleoprotein gene of influenza A virus (NP-A), nucleoprotein gene of influenza B virus (NP-B), and non-structural protein gene of influenza B virus (NS-B) were used for typing. Primers of the hemagglutinin glycoprotein gene of influenza A/H1N1 and A/H3N2 viruses (H1-A and H3-A) and neuraminidase glycoprotein gene of influenza A/H1N1 and A/H3N2 viruses (N1-A and N2-A) were used for subtyping purposes. These primers were designed from conserved and consensus regions of about 30 different relevant isolates retrieved from GenBank database using multiple alignments [Table - 1]. [13] Sequencing and Phylogenetic Analysis Twelve isolates were randomly selected for molecular characterization using bidirectional sequencing of HA1 gene from influenza-positive samples. They were subjected to PCR analysis of H1 and H3 gene followed by gene sequencing. The samples were labelled as: Shiraz 26, Shiraz 27, shiraz 32, Shiraz 33, Shiraz 2, Shiraz 5, Shiraz 13, Shiraz 29, Shiraz 37, Shiraz 54, Shiraz 6 and Shiraz 9. The H1 (543 bp) and H3 (292 bp) fragments from influenza A virus were used for sequencing and phylogenetic analysis. The segments were originally designed for multiplex PCR and performance does not cover all of the antigenic sites. However, it covers at least three out of 5 HA antigenic sites in both the strains. The PCR products of the HA fragment from H1 and H3 genes were purified by a commercial gel extraction kit (CoreBio® , Seoul, South Korea) and subsequently sequenced in both directions by a CEQTM 2000 Dye Terminator Cycle Sequencing kit (Beckman Coulter, Fullerton, CA) using an automated CEQTM 2000XL Microcapillary DNA Analysis sequencing Apparatus (Beckman Coulter). [13] About 50 H1 and H3 gene sequences were used in this study as reference to compare with the Iranian influenza H1 and H3 gene sequences. Alignment was performed using CLUSTALX software, version 1.81. Genetic distance was estimated by the Kimura two-parameter matrix. Phylogenetic trees were constructed by the neighbour-joining method; Bootstrap re-sampling and reconstruction was performed 1,000 times to confirm the reliability of phylogenetic trees. The computer software of the Molecular Evolution Genetic analysis (MEGA), version 2.1. [14] has been utilized in this study for phylogenetic and molecular evolutionary analysis and nucleotide differences within and between the isolate sequences. Nucleotide GenBank Accession Numbers The nucleotide sequences obtained in this study have been submitted to GenBank under the following accession numbers: EU430036, EU430037, EU430038, EU430039, EU430040, EU430041, EU430042, EU430043, EU430044, EU430045, EU430046, EU430047 Results Twenty-six samples were found positive by culture HI and HA assays. The RT-PCR based molecular typing and subtyping performed by using templates standard strains showed that 12 and 14 specimens were H1N1 and H3N2 influenza subtype respectively [Figure - 1]. No influenza type B was isolated in this study. These results were also consistent with HI assay. Sequence and Amino acid Analysis: Four of the 12 influenza A subtype H1N1 and eight of H3N2 influenza A subtype isolates were randomly selected for molecular characterization using direct nucleotide sequencing of the HA1 gene and compared with other sequences in Gen Bank especial current vaccine strains during 2005-2007 influenza seasons. Based on nucleotide alignments, Iran Fars province H1N1 isolates shared identical HA1 sequences with each other. Among Iranian H1N1 isolates maximum similarity (99-100%) was observed with the A/South Australia/55/2005 strain and 99% similarity with A/NewCaledonia/20/99 strain base on nucleotides and amino acids sequences [Table - 2]. The alignment of the amino acids in HA1 protein of 8 H3N2 isolates from Shiraz Iran with the A/California/7/2004 vaccine, indicated that 20 evident amino acid changes were present in most of the isolates [Table - 3]. Six of the eight strains exhibited novel asparagines to lysine, isolucine to leucine, and proline to serine substitution at positions 189,226 and 227 respectively [Table - 3]. These substitutions are noteworthy because mutation at position 189 alters the antibody-binding site B. Also mutation at positions 226 and 227 appears to marginally affect the HA surface molecule. These substitutions reside within the antibody-binding site. [15] Phylogenetic Analysis Genetic relationships of the HA1 region of the A/H1N1 [Figure - 2]a and A/H3N2 [Figure - 2]b isolates with vaccine strains and other influenza viruses were constructed by neighbour joining analysis with 1000 bootstrapped replicates. These analyses showed that Iranian A/H1N1 isolates were related to the NewCaledonia/20/99 vaccine strain and cluster in a unique branch [Figure - 2]a. Moreover, phylogenetic analysis showed that the Iranian A/H3N2 isolates make up a distinct branch in the evolution of H3N2 viruses when as compared with vaccine and reference strains [Figure - 2]b. Discussion The development of an effective influenza vaccine depends on strains of virus obtained from outbreaks of the disease. Annually, this is achieved by the World Health Organisation (WHO) through isolation of appropriate strains prevalent in different geographic regions around the world. In this connection, it is of considerable importance to detect minor antigenic changes (drifts) occurring in HA1 protein of viral hemagglutinin, a determinant crucial to the manufacture of vaccines. Antigenic specificities of viruses are determined by the number of modifications introduced into the vaccine formulation. [16] The occurrence of new antigenic changes in influenza viruses A/H1N1 and B is slow and gradual, compared with that of H3N2 which is a rapid process. According to WHO and CDC influenza weekly report 2005, the influenza vaccine used in northern hemisphere during 2005-06 contained H3N2 (A/California//7/9) and H1N1 (A/NewCaledonia/20/99) strains. Several studies have shown that H1N1 strain resembling A/Beiting/262/95 emerged in China during 1994-95 and spread across Asia within the space of 5 years. [17] Subsequent outbreaks of H1N1 similar to A/Bayern/7/95 occurred in other parts of the world from 1996 to 1999. In May and June 1999, A/NewCaledonia strain, a novel antigenic variant of A/Beijing/262/956, was first reported from NewCaledonia, southern pacific. [18] Shortly afterwards, A/NewCaledonia-like viruses were identified throughout Asia and South Africa which led to subsequent outbreaks in many countries during 2000-01. The continued presence of NewCaledonia-like viruses in global communities urged the WHO to use these H1N1 strains for preparation of seasonal influenza vaccines. [19] A new variant of the H3N2 influenza virus appeared in California during 2003-04. It superseded A/Fujian/H3N2 in 2004-05 and in February 2004 was adopted by the WHO as a constituent of the influenza vaccine for 2005-06. The substitution of new substrain of H3N2 is made relatively rapidly. This is evidenced by as many as 10 antigenic changes introduced into influenza vaccine in the period from 1986-1988. Certain strains of influenza viruses in Iran have already been demonstrated by serologic studies. The annual patterns of these isolates were similar to those reported elsewhere. They were A/Non Chang/933/95 in 1997-98, A/Sydney/05 in 1999-2000 and NewCaledonia in 2000-01. The seasonal prevalence of influenza viruses in Iran was found to be from November to April. The strains from seasonal outbreak in Shiraz during 2003-04 were further studied with respect to nucleotilde and amino acid sequences. Of the 24 isolates, 17 and seven strains were H1N1 and H3N2 respectively. The results obtained differed from those reported by CDC which showed the predominance of H3N2 over H1N1 subtypes. The study revealed that H1N1 isolates exhibited the greatest similarity (99%) to South Australia variant and bore a resemblance of about 98.51% to 99% to A/New Caledonia/20/99 vaccine strain. Considerable similarities were observed among H3N2 isolates which were antigenically nearest to Moscow strains. Nucleotide sequence analysis of these H3N2 variants demonstrated a genetic relationship to A/Fujian-like/411/02 vaccine viruses. The amino acid sequence analysis showed that H1N1 isolates differed from New Caledonia vaccine strains in four to eight amino acids. These amino acid substitutions were not located in antigenic sites. However, H3N2 isolates varied from Fujian vaccine viruses in only two to four amino acid in positions other than HA1 antigenic site. [13] With regard to the present study, of 26 isolates, 12 and 14 strains were H1N1 and H3N2 respectively. Nucleotide sequence analysis of 12 isolates randomly selected from 26 isolates showed that four were H1N1 and eight H3N2 subtypes. H1N1 isolates exhibited greatest similarity (99-100%) to A/Shiraz and about 99% resemblance to A/Victoria/504/2005 and A/South Australia/55/2005. A relationship as close as 98.4 to 99% was found between H1N1 isolates and A/NewCaledonia/20/99 vaccine strain. These subtypes were different from A/NewCaledonia/20/99 vaccine virus in only five amino acids whose substitutions were in regions other than distal portion of HA1 antigenic sites. This showed that they were of A/NewCaledonia/20/99-like generations and differed from A/NewCaledonia vaccine strain in 15 to 16 nucleotides. The nucleotide sequence of eight H3N2 isolates were studied with respect to information contained in the GenBank. Isolates 6 and 9 exhibited about 99% similarity to A/Georgia/4/2005 and A/Texas/13/2004 and also demonstrated 99% resemblance to A/California/7/2004 vaccine strain but diverged from the latter in only four amino acids. The substitution of these amino acids in regions other than HA1 antigenic site indicated that the isolates belonged to A/California/7/2004 generations. Isolates 2, 5, 6, 9, 13, 29,37 and 54 resembled A/Philippines/2/82 and A/Cheng-mei/4/85 by 98 to 99%. These isolates differed from A/California/7/2004 vaccine strain in 15 to 18 amino acids, but their similarity to the vaccine strain was not perceptible. Locations of 145, 155, 156, 189, 226 and 227 were active antigenic sites and any substitutions in these sites are epidemiologically important. Substitution of amino acid 189 was located in site B antigenicity, whereas replacing amino acids 226 and 227 was associated with site D antigenicity as shown in [Table - 3]. On antigenic sites 189, 226 and 227, the amino acids asparagines, isoleucine and proline were replaced by lysine, leucine and serine respectively. Isolates 2, 5, 13, 29,37 and 54 varied, in 26 to 32 nucleotides, from A/California vaccine strain used in the same year. These isolates bore a slight resemblance to A/California vaccine strain following amino acid substitutions in their HA1 antigenic sites. Other studies showed that strains with minor antigenic variations were of great epidemiologic significance. These strains exhibited two or more amino acid substitutions in at least two antigenic sites of HA1 molecule. [20] In 1992, the evolution of H3N2 was found to be associated with changes in conserved amino acids of receptor binding site particularly amino acid 226 which was considered to be the receptor specific epitope. This was situated close to timer faces of globular structure on D antigenic site at the top of HA1 molecule. Variations occurring in this region were associated with the ability of viruses to grow in mammalian and avian cells. [21] Phylogenetic studies of influenza viruses in 1982-2006 revealed two consecutive lineages of H1N1 and H3N2. H1N1 strains isolated in various regions around the world during 1988-1996 were considerably distant from those prevalent in 1999-2006 and formed a distinct evolutionary branch. [21] Similarly, H3N2 strains isolated during 1982-1993 were easily distinguishable from those circulating in 1999-2007. Also, phylogenetic analysis supported the genetic relationship between Iranian isolates and A/NewCaledonia vaccine strain and indicated that they evolved from A/NewCaledonia-like strains. However as shown by phylogenetic study, the six H3N2 isolates with notable antigenic changes differed from A/California vaccine strain [Figure - 2]b. In conclusion, several different subtype strains of the H3N2 circulate concurrently during 2005/7 influenza outbreaks in Shiraz. All these isolates demonstrated reasonable antigenic variation at least in two out of five important antigenic sites form A/California/7/2004 vaccine strain, which make this strain unsuitable for being used as an standard vaccine strain. In contrast, H1N1 subtypes isolates showed a notable antigenic and sequence resemblance to A/NewCaledonia/20/99 vaccine, hence this vaccine strain predicted to be capable of conferring sufficient immunity against circulating H1N1 subtypes in population. Acknowledgments This work was financially supported by a grant (84-2399) from Vice Chancellor for Research, Shiraz, University of Medical Sciences. The authors are grateful to Dr. Elena percivalle (Servizio di Virologia, IRCCS Policlinico San Matteo, Via Taramelli 5, 27100 Pavia, Italy) for her critical review of the paper. References
Copyright 2010 - Indian Journal of Medical Microbiology The following images related to this document are available:Photo images[mb10036f1.jpg] [mb10036t2.jpg] [mb10036f2.jpg] [mb10036t3.jpg] [mb10036t1.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}